Small Molecular Weight RNAs:
Nuclear structural features;
Nucleus is the heart of eukaryotic cells. It is a membrane bound cell organelle. It has chromosomes which contain genomic information in its heritable DNA. Nucleus encodes several important structures and functions. Thus it contain several structure, but none of the demarked by membrane envelops. Nuclear architecture depending upon organization and components vary cell type to cell type.
Neat RNA is an architectural RNA contains scaffolds, contains Neat RNA that produces Para speckles. It is important to understand what are the particulates found and their structural features and functions; a brief account is given below. Eg; pluripotent stem cells, human ES cells, cancer cells and many others.
Some of the sub nuclear bodies found in the nucleus:
Nucleolus; Cajal bodies-0.2-2.0um, PIKA- 5um, PML bodies- 0.2-1.0um, Speckles and Paraspeckles-0.2-2.0um and Speckles-20-25nm.
|
Subnuclear structure sizes |
|
|
|
Structure name |
Structure diameter |
|
|
Cajal bodies |
0.2–2.0 µm |
|
|
PIKA |
5 µm |
|
|
PML bodies |
0.2–1.0 µm |
|
|
Paraspeckles |
0.2–1.0 µm |
|
|
Speckles |
20–25 nm |
|
Polymorphic interphase karyosomal association (PIKA), Promyelocytic leukaemia (PML) bodies, Paraspeckles, and Splicing speckles.

Jeanne Lawrence-UMass Medical School; Jeanne Lawrence-UMass Medical School; http://www.umassmed.edu/
Cajal bodies: They were first discovered by Santiago Ramon Cajal in 1903. They are spherical sub organelles found mostly in proliferative cells, such as cancer and embryonic cells. They are not encircled by any membranes; they mostly contain RNAs and coiled proteins. They are coiled structure, a marker protein found was p80/coilin. They are involved in RNA mediated metabolic processes. RNA processing specifically small nucleolar RNA (snoRNA) and small nuclear RNA (snRNA) maturation, and histone mRNA modification. They are found in animal cells, plant cells and yeasts cells. These bodies are involved in modification and assembly of U snRNP. Similar to cajal bodies one finds another similar structure called GEMS (Gemini of Cajal Bodies). They do contain a protein called survival of motor neuron (SMN) whose function relates to snRNP biogenesis. Gems are found to be involved in assisting CBs in snRNP biogenesis, Jeanne Lawrence-UMass Medical School; http://www.umassmed.edu/
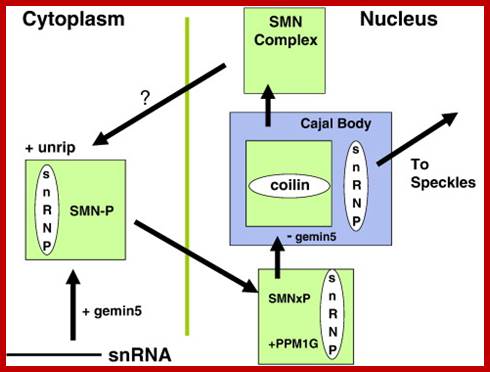
The Spliceosomal U snRNP Cycle. Newly-synthesized spliceosomal U snRNAs (pink squares) are exported to the cytoplasm where they are assembled into U snRNPs (red squares) by the SMN complex, which also transports them into the nucleus and delivers them to the Cajal body. Further modifications of U snRNPs and assembly of the tri-snRNP occurs in the Cajal body before the snRNPs are assembled into the spliceosome in situ on newly-transcribed pre-mRNA in the perichromatin fibrils (PF) at the chromatin periphery. Essential splicing factors are supplied by the splicing speckle, or interchromatin granule (ICG). After each splicing step, UsnRNPs are re-cycled to the Cajal body for re-assembly and the spliced mRNA with attached proteins (mRNP) is exported to the cytoplasm. See text for references. Inset A: This model of the SMN complex is based on data in Ref. [121]. The balls represent SMN (“1” = gemin1), gemins 2–8 and unrip. The Sm ring is the U snRNA with seven Sm core proteins attached. Glenn E. Morrishttp://www.sciencedirect.com/
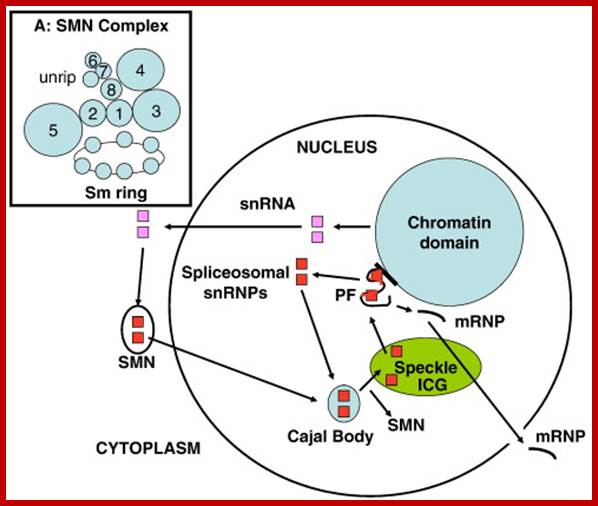
The SMN (Survival of Motor Neurons) complex cycle. In the cytoplasm, the U snRNP core is assembled by the SMN complex, which includes the WD-repeat proteins, gemin5 and unrip. Gemin5 delivers the snRNA and SMN is maintained in a highly-phosphorylated state (SMN-P). Methylation of the snRNA by the TGS1/PIMT methylase enables its transport to the nucleus, where SMN is dephosphorylated by PPM1G before it binds to coilin in the Cajal body. SMN complexes without gemin5 or snRNPs remain in the Cajal body and the U snRNPs undergo further modification and assembly before they are released to transcription sites for spliceosome assembly, picking up splicing factors from the “speckles” on the way. See text for references; Gems are the nuclear site of accumulation of survival motor neurons (SMNs), an insufficiency of which leads to the inherited neurodegenerative condition, spinal muscular atrophy (SMA). Gems are the nuclear site of accumulation of survival motor neurons (SMNs), an insufficiency of which leads to the inherited neurodegenerative condition, spinal muscular atrophy (SMA). Glenn E. Morrishttp://www.sciencedirect.com/
Nuclear speckles: a model for nuclear organelles: They sub nuclear structures and they are enriched in pre-mRNAs and splicing factors. They are located at inter chromatin regions. RNAs and RNA-Proteins recycle continuously between speckles and nucleoplasm. Sometimes referred to as interchromatin granule clusters or as splicing-factor compartments, speckles are rich in splicing snRNPs. Speckles are dynamic structures; their size, shape, and number vary, among different cell types, according to the levels of gene expression and in response to signals that influence the pools of active splicing and transcription factors available. They contain splicing factors

Speckles form in the interchromatin space. HeLa cells showing splicing factors localized in a speckled pattern as well as being diffusely distributed throughout the nucleoplasm. Bar = 5 µm. https://www.ncbi.nlm.nih.gov
PML Bodies:
The PML gene and NBs suppress tumors. The PML ge foctic leukemi (APL).und in the in premyelocyte of pateints suffering from Acute promyelone was first identified as the fusion partner of retinoic acid receptor alpha alpha RAR at the break point of common chromosomal translocation. PML bodies are DNA damage sensors. http://www.dellairlab.medicine.dal.ca
They are more or less spherical in shape; they contain promyelocytic leukemia protein (PML). They are in association with Cajal bodies and cleavage proteins. They are found associated with nuclear matrix and regulate many functions DNA replication, transcription and or RNA processing and epigenetic silencing.
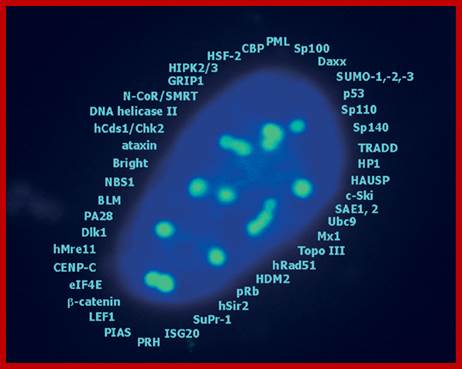
A list of cellular proteins found (identified) in PML-NB; they contains components for transcriptional regulation , maintenance of genomic stability, post translational protein modification, protein degradation , DNA repair and apoptosis. http://www.narture.com, G Hofmann and H Will
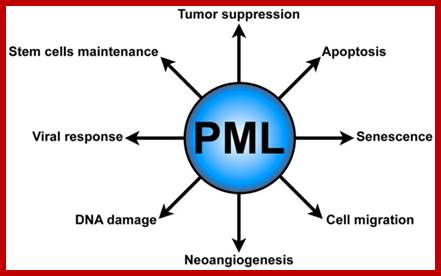
Promyelocytic leukemia bodies (PMLs) functions. PML plays several critical cellular functions, such as apoptosis and senescence, neo-angiogenesis, cell migration, DNA damage response, antiviral defense, and hematopoietic stem cell maintenance.journal.frontiersin.org
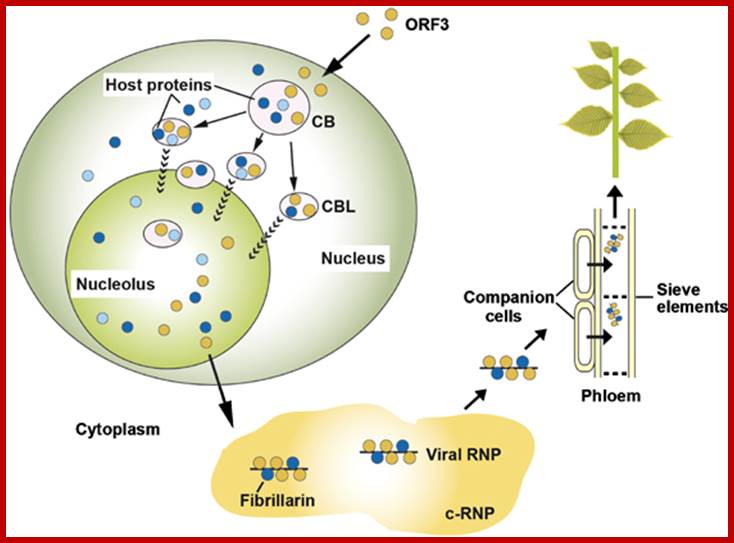
Model of GRV infection and the role of ORF3 protein, fibrillarin, CBs and the nucleolus. Upon GRV infection, the ORF3 protein is produced in the cytoplasm and is targeted to CBs reorganizing them into multiple CBLs. The CBLs then move to and fuse with the nucleolus by an unknown mechanism. Host proteins are likely to be involved in targeting of the ORF3 protein to the CBs, reorganizing CBs and causing their fusion with the nucleolus. One such host protein is fibrillarin and the ORF3 protein causes the relocalization of some of the nuclear/nucleolar fibrillarin pool to the cytoplasm where viral RNPs containing the ORF3 protein, fibrillarin and viral RNA accumulate. When produced in companion cells, the viral RNPs are able to migrate into the phloem sieve elements where they are transported to the rest of the plant to generate a systemic infection. Orange circles—the ORF3 protein; dark blue circles—fibrillarin, light blue circles—other host proteins including coilin and U2B".Sang Hyon Kim, Eugene V Ryabov, Natalia O Kalinina, et al

A slice through the nucleus of a cancer cell grown in culture. The cell is stained with markers that reveal different sub-nuclear structures such as DNA, splicing speckles, nucleoli and paraspeckles. Specific molecules are held in the paraspeckles, preventing them being made into proteins. Dr.Archa Fox WAIMR
Nuclear speckles: a model for nuclear organelles: They sub nuclear structures and they are enriched in pre-mRNAs and splicing factors. They are located at inter chromatin regions. RNAs and RNA-Proteins recycle continuously between speckles and nucleoplasm. Sometimes referred to as interchromatin granule clusters or as splicing-factor compartments, speckles are rich in splicing snRNPs. Speckles are dynamic structures; their size, shape, and number vary, among different cell types, according to the levels of gene expression and in response to signals that influence the pools of active splicing and transcription factors available. They contain splicing factors

Speckles form in the interchromatin space. HeLa cells showing splicing factors localized in a speckled pattern as well as being diffusely distributed throughout the nucleoplasm. Bar = 5 µm. https://www.ncbi.nlm.nih.gov
There are different types like splicing speckles, paraspeckles
The other forms of speckles were discovered by Fox et al. in 2002. Paraspeckles are irregularly shaped compartments in the nucleus' interchromatin space; the "para" is short for parallel and the "speckles" refers to the splicing speckles to which they are always in close proximity. They are found to act as storage structures for splicing factors.
Another type of speckles called Paraspeckles are irregularly shaped and they are dynamic structures. They are associated with proteins such as PSP1, p54nrb, PSP2, CFI(m)68, and PSF, they form a crescent shaped peri-nucleolar cap in the nucleolus.
Polymorphic interphase karyosomal association structures PIKA. Antibodies recognize such structures, which change their structural features during cell cycle. Yet its structure and functional features are not determined.

Cancer gene regulation; Dr Archa Fox
They are transcription dependent and in the absence of RNA Pol II transcription, the paraspeckle disappears. When observed by immunofluorescence microscopy, they usually appear as 20–50 irregularly shaped structures that vary in size. Speckles are dynamic structures, and their constituents can exchange continuously with the nucleoplasm and other nuclear locations, including active transcription sites. Studies on the composition, structure, and dynamics of speckles have provided an important paradigm for understanding the functional organization of the nucleus and the dynamics of the gene expression machinery.
Non Coding but Functional ncRNAs ;
These RNAs are transcribed from genomic DNA but not translated into proteins. They are epigenetic related ncRNAs such as miRNA, siRNA, piRNA and LncRNA. They are involved in gene expression and post transcriptional level. They can be divided into two mai groups ; short ncRNA size 30 ntds, and long ncRNA >200 ntds or more. The short nc RNAs are miRNAs and si RNAs and piwi-interacting RNAS (piRNAs). They are found to play important role in chromatin- heterochromatization, histone modification and DNA methylation and gene silencing. NEAT RNAs-Nuclear Enriched Abundant Transcript RNAs , long ~3.2 kb long. Neat is often induced in mouse brains during infection by Japanese encephalitis virus and Rabies. Often one can observe these RNAs as constitutively expressed ribose nucleic acids in non-neuronal tissues. They often localized in paraspeckles. Another RNA called MALT1 RNA is non coding RNA about 53-61 ntd long. They are processed from long nc RNAs. They show similarity with tRNAs. Often one finds they are upregulated in malignant cancer cells. This RNA is speculated in regulating alternative splicing of precursor mRNAs.

http://www.whatisepigenetics.com/
Most of the RNA are named as to their functions such as rRNAs, tRNAs, mRNAs, snRNAs, scRNAs, snoRNAs, scaRNAs, few antisense RNAs and few more (interesting) RNAs. In recent years many small molecular wt RNAs have been discovered and the list is still increasing. However, note that mRNAs and tRNAs have codon and anticodon information respectively; yet can we call tRNAs as noncoding RNAs?
The Human Genome Project (HGP) 1990-2003 and ENCODE between 2001-2012 has opened a new vision in microcosmic of nature of DNA structure and function that pervades the whole cell into vibrant and dynamic mass of molecules that perform functions what we call it as life. Ewan Birney (who?) lead analyst and coordinator of ENCODE ‘Encyclopedia of DNA Elements” project says “, I think it’s going to take this century to fill in all the details. That full reconciliation is going to be this century’s science.” Work on ENCODE encompassing 442 dedicated scientists on 147 and odd cell types employing 24 types of experiments for ten long years was published simultaneously in 30 journals, coordinated, open-access papers in Nature, Genome Research and Genome Biology as well as in Science, Cell, JBC and few more.

ENCODE: Encyclopedia of DNA Elements

https://www.encodeproject.org/
The ENCODE (Encyclopedia of DNA Elements) Consortium is an international collaboration of research groups funded by the National Human Genome Research Institute (NHGRI). The goal of ENCODE is to build a comprehensive parts list of functional elements in the human genome, including elements that act at the protein and RNA levels, and regulatory elements that control cells and circumstances in which a gene is active.
ENCODE often called the human Encyclopedia of DNA Elements (Nature), the printout of “Dark Matter” of our genome that could run” 30 kilometers long” (1000bp per square cm). According to ENCODE analysis, >80 percent of the genome has biochemical functions. That’s where we are now: a comprehensive 3-D portrait of a dynamic, changing entity, rather than a static 2-D map.
Some authors express that 98% of the human genome sequence is transcribed (encoded information), some are skeptic about this analysis as noncoding DNA. It is defined as all of the DNA sequences within a genome that are not found within protein-coding sequences, and they are not represented as amino acid sequences of expressed proteins. By this definition, more than 98% of the human genome is comprised of non-coding DNA (ncDNA).
There are criticisms, but and yet it is a remarkable expose’ of our DNA both coding and noncoding. To include all such sequences within the bracket of “functional” sets a very low bar. Michael Eisen from the Howard Hughes Medical Institute said that ENCODE’s definition as a “meaningless measure of functional significance” and Leonid Kruglyak from Princeton University noted that it’s “barely more interesting” than saying that a sequence gets copied (which all of them are). And Michael White from the Washington University in St. Louis said that the project had achieved “an impressive level of consistency and quality for such a large consortium.” He added, “Whatever else you might want to say about the idea of ENCODE, you cannot say that ENCODE was poorly executed”, it is nonsense, instead of appreciating.

Four experimental approaches (A–D) to identify candidates for ncRNAs are shown. (A) Identification of ncRNAs by chemical or enzymatic sequencing of extracted abundant RNAs. (B) Identification of ncRNAs by cDNA cloning and sequencing; three different methods are indicated to reverse transcribe ncRNAs, usually lacking poly(A) tails, into cDNAs (e.g. by C-tailing, C-tailing and linker addition, or linker addition, only, followed by RT–RCR). (C) Identification of ncRNAs by micro-array analysis. DNA oligonucleotide covering the sequence space of an entire genome are spotted onto glass slides, to which fluorescently labelled samples, derived from cellular RNA, is hybridized. (D) Identification of ncRNAs by genomic SELEX. By random priming, the sequence of a genome is converted into short PCR fragments containing a T7 promotor at their 5′ ends. Subsequently, in vitro transcription by means of T7 RNA polymerase converts this genomic sequence of an organism into RNA fragments, which can then be assayed for function, such as binding to a specific protein or small chemical ligand, by SELEX. Alexander Hüttenhofer* and Jörg Vogel1
http://nar.oxfordjournals.org/
A list of small Mol.wt RNA (incomplete):
Except mRNAs and tRNAs for they have coding and anticodon information; all other RNAs can be considered as noncoding RNAs. HUGO gene nomenclature committee has identified~ 3000 such genes. Rfam is a comprehensive collection of data on non-coding RNAs. RFAM identifies 13,400 candidates of ncRNAs belonging to 172 families. Among the noncoding RNAs except rRNAs (structural) and long noncoding-lncRNAs (200 or more ntds long), all others are small mol.wt RNAs. Besides the said species of ncRNA there are more RNAs such as Aptamer RNAs, Transacting RNAs, mi/siRNAs, Xist RNA, tsixRNA, Tm RNA, Srp RNA, antisense RNA, lincRNA, Vault RNA, YRNA, Rasi RNA, TasiRNA, DsR RNA, Pi RNA, XnC RNA, Scan RNA, SRNA, tiRNA, LincRNA, stRNA, SncRNA, Sca RNA, SnoRNA, SnRNA, UsuRNA, StRNA, Rhyb RNA, OxySRNA, ISI RNA, pRNA, Activator RNA, SRA RNA, AtRNA, EfferenceRNA, Telomeric RNA, RNaseP RNA, Riboswitch RNA, PrimerRNA, tiRNA, Srp RNA, M1RNA, MRP-RNA, tmRNA, 7LSKRNA, 7SLRNA, 4.5SkRNA, 21URNA, ShRNA, Si/MiRNAs, HOTAir RNA, TSSa RNA, scnRNA, ASRNAs, lsiRNA, natsiRNA, lsiRNA, PASRs, aTASR RNAs, TSSaRNA, RERNA, uaRA, X-ncRNA, huYRNA, usRA, NF-RNA (snaRs RNA), T-UCRRNA, GRCRNA, bifunctional RNAs (SgrsRNA), RNAIII, SRA, VegtRNA, Oskar RNA, SR1RNA, Malt RNAs, OLE RNAs, Ribozyme RNA, group intron-ribozymes and few more and many more, perhaps; the next decade will be of NC RNA decade, heralding the era of RNomics similar to Genomics, Proteomics, Transcriptomics and Metabolomics; the said RNomics is very important. It is presumed it will be the decade of small Molecular weight RNAs ahead of information and experiments.
The ncRNAs have been coded for by specific genes and some are spliced out intronic segments. They have very important roles in regulation gene expression at transcriptional and translational level. They play roles in response to stress, in response developmental signals.
It is now known in human genome 3.3 x 10^9 bp, only 1 or 1.2% of the genome is involved in coding proteins i.e. the region that is translated as amino acids. The number of protein coding genes once thought to be 1.5 million has now been reduced to 34000, which is now further reduced to ~22000. Now it is realized that what the rest of the genomic DNA doing. Some of these NC RNA are coded for by many hundreds of genes, some transcribed by RNA Pol-II and some by RNA pol-III and some are derived from the transcripts such as spliced out Introns, perhaps account for the rest of the genomic not involved in coding for proteins. The term useless DNA or junk DNA once very popular now looks like that the term should be discarded. Interestingly human genome contains a lot of DNA in the form of molecular fossil and variety of inactive transposonic DNA. Does this excess DNA that is of no use has any functions? Yes they absorb point mutations and even some non homologous recombination and not properly repaired DNA damage.
Numerous classes of noncoding DNA have been identified, including genes for noncoding RNA (e.g. tRNA and rRNA), pseudogenes, and introns, untranslated regions of mRNA, regulatory DNA sequences, repetitive DNA sequences, and sequences related to mobile genetic elements. Untranslated components of components of protein-coding genes (e.g. introns, and 5' and 3' untranslated regions of mRNA) included.
Bacterial noncoding RNAs are referred to as sRNA for they are short RNAs. Work on nitrogen fixing bacteria Sinorhizobium meliloti strain 2011, lead to the prediction of 6308 CDS as well as 1876 ncRNAs; recent findings that antisense transcription activity is widespread in bacteria. Moreover, 4077 TSSs upstream of protein-coding or non-coding genes were precisely mapped providing valuable data for the study of promoter regions. E.coli contains a variety of ncRNAs involved in transcription and translation regulation.
In eukaryotes the Telomeres and Centromeres and other noncoding regions serve as Origins of DNA replication. Finally several regions are transcribed into functional noncoding RNA that regulate the expression of protein-coding genes (for example), mRNA translation and stability (see miRNA), chromatin structure (including histone modifications, for example), DNA methylation for example.
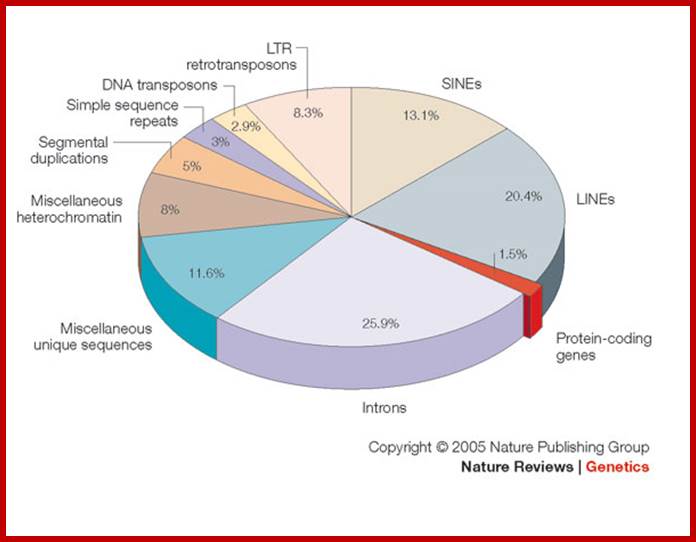
Pseudogenes in the human genome are in the order of 11224-13,000 (of which 863 are associated with active and inactive chromatin); in some the number of pseudogenes is same as protein coding genes. The human genome contains genes encoding 18,400 (?) ncRNAs, including tRNA, ribosomal RNA, microRNA, and other non-coding RNA genes. Introns, 5’ n 3’ UTRs, Mobile elements within the human genome can be classified into LTR Retrotransposon (8.3% of total genome), SINEs (13.1% of total genome) including Alu elements, LINEs (20.4% of total genome), and Class II DNA transposons (2.9% of total genome).
They are involved in DNA replication (YRNA), Gene regulation, Transacting RNAs, microRNAs, RNase P, 5S rRNA, SRP RNA, and U6 snRNA, 7SK RNA, 6sRNA, OxyS RNA, cis acting ncRNAs,some act as genome defence RNAs, some are found in chromosome structure and modification (Xist RNA), Tsix RNA, bifunctional (dual functional) RNA and many others.
Annotated small RNAs:
Currently, a total of 13,400 small RNAs are annotated by ‘Gencode’, 85% of which correspond to major classes: small nuclear (sn)RNAs, small nucleolar (sno) RNAs, RNAi-micro (mi)/Si RNAs, transfer (t)RNAs, (promoter-associated short RNAs (PASRs) and termini-associated short RNAs (TASRs), and their position relative to TSSs and transcription termination sites is similar to previous results. OSKAR RNAs, ENCOD40 RNA, SR1 RNA, sgrS RNA
1. The long noncoding RNA has exceptional features and biotypes with respect to protein coding genes. Antisense RNAs: Locus that has at least one transcript that intersect any exon of a protein-coding locus on the opposite strand, or published evidence of antisense regulation of a coding gene.
2. LincRNA: Locus is intergenic non-coding RNA loci.
3. Sense overlapping: Locus contains a coding gene within an intron on the same strand.
4. Sense intronic: Locus resides within intron of a coding gene, but does not intersect any exons on the same strand.
5. Processed transcript: Locus where none of its transcripts contain an open reading frame (ORF) and cannot be placed in any of the other categories because of complexity in their structure.
In summary the lncRNAs data set in GENCODE 7 consists of 5,058 LincRNA loci, 3,214 antisense loci, 378 sense intronic loci and 930 processed transcripts loci. LncRNAs (lnc) display a striking bias toward two-exon transcripts, they are predominantly localized in the chromatin and nucleus, and a fraction appear to be preferentially processed into small RNAs.
Up to 450 000 non-coding RNAs (ncRNAs) have been predicted to be transcribed from the human genome Mathieu Rederstorff, Stephan H. Bernhart etal.; such genes are believed to be associated with specific TFs.
In Eukarya, most if not all known ncRNAs are associated with RNA binding proteins thus forming ribonucleo-protein particles or RNPs. lncRNAs display a striking bias toward two-exon transcripts, they are predominantly localized in the chromatin and nucleus, and a fraction appear to be preferentially processed into small RNAs.
Synergy between sequence and size in Large-scale genomics;T. Ryan Gregory; http://www.nature.com/
As a prime example, the human genome might contain as few as 20,500 genes, comprising less than 1.5% of the total genome sequence. Most of ncRNA coding regions are found in introns, and they are found as pseudo genes, transposable elements (SINEs LTR, LINEs and MITEs). Introns and transposable elements account for 26% and 45% respectively.
|
Type of RNA |
Genes |
Size in Nucleotides |
characters |
location |
functions |
|
rRNAs |
rRNA |
Vary from 120->2800 |
NC |
Ribosomes |
Structural organization and translation |
|
tRNAs |
tRNA |
65-85 |
Folded clover leaf |
cytoplasm |
Transfer aa n decoding |
|
SnRNAs |
Sn RNA |
120- > 300 |
Sec.structures |
Nuclear sap |
Processing premRNAs |
|
ScRNAs/ SrpRNA |
scRNAs |
>180 |
Sec. structure |
cytoplasm |
Positioning of ribosomes on to ER |
|
Sno-RNAs |
snoRNAs and Sno RNA genes |
Variable 87-275ntds |
No cap |
Nucleolus |
Methylation& pseudouridination-n of rRNA |
|
Sca RNAs |
Sno RNA U85 |
120->200 |
Sec. structures |
Cajal bodies |
Processing snRNAs and mRNAs |
|
RNAi and mi/si RNAs |
SiRNAs ,MiRNAs |
22-24 ntds long |
Single stranded |
Nucleoplasm/ cytoplasm |
Block mRNA translation or/ and induce destruction mRNA |
|
RNAs with Riboswitches |
Small mRNAs |
Can be as large as mRNAs |
mRNAs-5’UTR and 3’UTR |
Nucleoplasm/ cytoplasm |
Bind to certain factors and regulate gene expression |
|
Antisense RNAs
|
Small RNAs |
Can have different sizes |
Single stranded |
Nucleoplasm/ cytoplasm |
Bind to mRNA and prevent it translation/savor-flavor tomato |
|
tm RNAs |
tmRNA |
250->363 |
Single stranded |
cytoplasm |
Completing translation of mRNAs without TER codon |
|
gRNAs |
gRNA |
60 -120 |
Single stranded |
mitochondria |
T.brucei Editing mRNAs |
|
Efference RNAs
|
NC genes |
|
Single stranded |
Nucleus/cytoplasm |
Regulation of Transcription and translation |
|
7.5 K RNA |
|
331ntd |
Single stranded |
nucleus |
Inhibits transcription elongation factor |
|
E,coli |
|
184 |
ss |
E.coli |
Promoter-transcriptional |
|
SRA |
|
875 |
|
human |
Steroid receptor oactivator |
|
Xist RNA |
|
16500 |
ss |
human |
x-chromosome inactivation |
|
At RNA |
|
100 000 |
|
|
hu autosomal gene imprinting-silncing |
|
Tel RNA |
|
451 |
|
hu |
Telomerase RNA,expansion of TEL DNA |
|
RNase P |
|
311 |
ss |
E.coli |
5’ endonuclease liberate 5 end of tRNA |
|
U2 snRNA |
|
186 |
|
hu |
Cis spicing |
|
|
|
|
|
|
|
|
RhyhB sRNA |
|
80 |
|
|
mRNA degration |
|
Oxy S RNA |
|
109 |
|
|
Repress translation, prevent ribosome binding |
|
DsrA sRNA |
|
87 |
E.coli |
|
Activate translation |
|
4.5s RNA |
|
114 |
|
|
Srp-prot |
|
Tasi RNA |
Transacting interfering RNA |
21 |
|
|
Convert 21ntd RNA into dsRNA |
|
rasiRNA |
|
24-26 |
|
Act on dsRNA |
Repeat associated short interfering RNA |
|
ScanRNA |
|
29 |
|
|
siRNA RNA interference |
|
LsiRNA |
Long siRNA |
30-40 |
|
|
Develop resistant to infection |
|
pRNA |
|
50-80 |
|
cytoplasm |
As scaffold promoter |
|
Activator RNA |
ss |
2.2kb |
|
|
Activation or repression |
|
Linc RNA |
|
|
|
nucleus |
Gene silencing |
|
SEA |
|
875nts |
|
|
coactivator |
|
Guide RNA (CYbg) |
|
68ntd |
|
|
mRNA editing |
|
RhyB sRNA |
|
80 |
|
|
mRNA degradation |
|
DsRA sRNA |
87 |
|
|
|
Activates over come inhibition |
|
tmRNA |
|
363 |
|
|
Adds tags to protein |
|
6sRNA |
|
|
|
Binds sigma 70 |
repression |
|
SeCIS |
|
|
|
|
|
|
|
|
|
|
|
Self splicing |
|
|
|
|
|
|
Self splicing |
|
|
|
|
|
|
Self splicing |
|
Sno RNA |
|
|
|
|
Alternate splicing-serotinin receptor RNA |
|
7SK RNA |
|
|
|
|
Negative regulator of RNAPII pTEFb |
|
6SRNA ECoi |
|
|
|
|
Binds RNAP –sig70, represses during sytationary phase |
|
OxyS RNA |
|
|
|
|
Binds to shine Delgarno- block translation |
|
B2 RNA |
|
|
|
|
RNAP III transcript, blocks transcription –heat shock |
|
|
|
|
|
|
|
|
PiRNA |
Piwi interacting RNA |
|
|
|
Block transcription of retrovirus |
|
RoX RNA |
RNA on the X |
|
|
|
epigenetic |
|
Bifunctional RNAs |
Sgrs, SRA,VegTRNA,Oskar, ENCOD |
|
|
|
|
|
|
|
|
|
|
|
|
Group1 RNA |
|
|
|
|
|
|
Group II RNA |
|
|
|
|
|
|
Group III RNA |
|
|
|
|
|
|
6sRNA |
|
|
|
|
|
|
|
|
|
|
|
|
|
B2 RNA |
|
|
|
|
|
|
50swRNAs |
|
|
|
|
|
4.5sRNA- 4.5S RNA, 94nts, is encoded by hundreds of tandemly linked genes, has a short half-life, and is hydrogen bonded in vivo to poly(A)-terminated RNAs in the cytoplasm of cultured mouse cells. In Ascites cells found to hybridize with 18S rRNA and mRNAs; it is 87ntd long. It has UUCCUUCCUU at its 3’ end which is homologous to 18s rRNA. Another 10ntd is complementary to 5.8s rRNA. Thus it indicates its regulatory role. Some are derived from intergenic region and three exhibit cis-antisense sRNAs. Deep sequencing reveals as-yet-undiscovered small RNAs in Escherichia coli; Atsuko Shinhara1,2, Motomu Matsui2, Kiriko Hiraoka, Wataru Nomura3, Reiko Hirano1, Kenji Nakahigashi1, Masaru Tomita, Hirotada Mori and Akio Kanai .
Another class 4.5s RNA and Ffh together in bacteria, form the signal recognition particle (SRP), which binds to the signal sequence and targets the nascent protein to the SRP receptor, FtsY, The SRP is involved in integration of nascent proteins into the membrane.
Short Interfering RNAs in Arabidopsis; Besides functioning as short siRNAs (by binding to mRNA and degrading the same), some long si RNAs bind to DNA sequences and methylate them and regulate the expression- Andrew Hamilton2,3, Olivier Voinnet2,3, Louise Chappell1 and David Baulcombe.
DsrA RNA: In E.coli double stranded regulatory RNA known forming a nanostructure. This RNA is 87ntds long in E.coli self assembles into nanostructures through complementary antisense structures. It regulates both transcription silencing and translation by promoting efficient translation of RpoS sigma factor under stress conditions. Three stem loops have different but specific functions.

DsrA RNA; http://en.wikipedia.org/
DsrA RNA 85nts long, activates the translation of Rpo proteins and suppresses H-NS protein, but it requires the Hfq (it acts as chaperone) protein for its function.
In E.coli itself there are 100 or more noncoding SMW RNAs. Deep sequencing has provided information about bacterial RNA of 50-200ntd long
The above list is incomplete, for there are RNAs called transacting RNAs, shRNAs, XistRNAs, stRNAs, LincRNAs and many more.
Sn and Sc-RNAs and Their Specific Features:
In late 70s and early 80s, identification of a disease called Systemic Lupus Erythematoses (SLE), led to the identification of a class of RNAs called small molecular weight RNAs and also their associated proteins. SLE is an autoimmune disease where antibodies are generated against their own nuclear proteins. The proteins are found to be associated with a set of small molecular wt. RNAs. Based on the sequences or sequence enrichment of a particular type, they are named as, for example U RNAs, where the RNAs are rich in U residues. They are also named as Sn RNAs and Sc RNAs for small molecular weight nuclear RNAs and small molecular weight cytoplasmic RNAs respectively. The total concentration of Sn RNAs is about one million per cell and their associated proteins is about 10^8 per cell. Concentration of sc RNAs and their proteins as Srps, though not clearly established, their number could be substantial, for they are involved in loading translating machinery on to endoplasmic reticular membrane surface.

BioMedCentral; http://www.springerimages.com/Images
|
Name of the RNA |
Type of RNA |
Size in ntds |
5’feature |
Location |
Function |
|
|
|
|
|
|
|
|
U1-RNA |
Sn-RNA |
165 ntds |
M,2,2,7 GpppN |
Nucleus |
Splicing
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
U2-RNA |
Sn-RNA |
188 ntds |
m2,2,7GpppN |
Nucleus |
Splicing |
|
U3-RNA |
Sno-RNA |
210 ntds |
M2,2,7GCapped? |
Nucleolus |
rRNA splicing |
|
U4-RNA |
Sn-RNA |
142 ntds |
M2,2,7GpppN |
Nucleus |
Splicing |
|
U5-RNA |
Sn-RNA |
116 ntds |
M2,2,7GpppN |
Nucleus |
Splicing |
|
U6-
RNA |
Sn-RNA
|
107 ntds |
CH#7pp-
H3ppp—
|
Nucleus
|
Splicing,
|
|
U7-RNA |
Sn-RNA |
56 ntds |
2’2’7’cap |
Nucleus |
Splicing of Histone mRNA |
|
|
ScRNAs |
scRNA |
~280 300 |
No caP |
cytoplasm |
|
|
|
4.5s-RNA |
bacterial |
94 ntds |
? |
? |
transport |
|
|
7SL-RNA |
Sc-RNA(srp) |
295 ntds |
No cap |
Cytoplasm, srp RNA |
Protein synthesis |
|
|
U11 RNA |
snRNA |
|
|
|
Plants/Similar to U1 RNA |
|
|
U12 RNA |
snRNA |
|
|
|
Plants/ Similar to U2 RNA |
|
Sn/Sc RNA genes:
· Most of the Sn or Sc RNA genes are smaller and they are found distributed among all chromosome. They are in clusters.
· Each of the Sn RNA genes exists in multiple copies ranging from 50 or to 200 or more copies each.
· Overall number of them can be a million or more.
· Most of them are in a complex called spliceosomes. They aggregated in a in small speckles within nuclear sap.
· In spite of large numbers, majority of them are nonfunctional and remain as pseudo genes. The ratio between pseudo genes and true (working) genes is 10:1. Most of the pseudo genes vary from true genes just by one nucleotide or so at 3’ end.
· Pseudo genes are often flanked by 6 to 20 bp perfect direct repeats.
· Pseudo genes are believed to be inserts of their own or of the same kind with in a functional gene.
· Many a times some processed genes by selective insertions become pseudo genes.
· RNA-pol II transcribes most of these genes and except U6 which is transcribed by pol-II and also by RNAPIII.
· The 5’ end of them is capped except U3, U6 and U7. The 3’ is terminated and processed by using C-Box.
· Monomethyl U1, U2, U4,U5 snRNA are transported out of the nucleus and they are subjected hypermethylation-at 2’2’7’ GpppX-,
· They are then get associated with the core group of six sm Proteins as rings, then are transported back into the Cajal bodies found in the nucleus.
The following table shows the location of sn U- RNA genes on specific chromosomes in Arabidopsis thaliana, 2n=10.
|
Chromosome |
U1 |
U2 |
U3 |
U4 |
U5 |
U6 |
Minor RNAs |
|
Chromo 1 |
3 |
3 |
5 |
2 |
7 |
2 |
2 |
|
Chromo 2 |
- |
3 |
0 |
1 |
- |
1 |
- |
|
Chromo 3 |
2 |
5 |
1 |
3 |
4 |
3 |
- |
|
Chromo 4 |
4 |
1 |
0 |
3 |
1 |
4 |
1 |
|
Chromo 5 |
6 |
6 |
5 |
3 |
3 |
3 |
1 |
Note: U2 and U5 clusters are found in chromosomes 1, 3 and 5
· Sn or Sc RNA gene promoter elements look more or less similar to protein coding genes; few snRNA genes that are transcribed by RNAP II lack TATA box but associated with upstream sequence elements and transcribed by RNAP II and its associated TFs.
· The Sn RNA6 is transcribed by RNAP III and the promoter contains TATA box, but associated with RNAP III associated TFs.
Small Mol.Wt Cytoplasmic RNAs:
The Sc RNAs produced are processed and soon they associate with nuclear proteins to form a complex as SRPs and move out into cytoplasm. Concentration of SRP complex is about 10^6 particles per cell. A classical example for sc RNA is 7sL RNA and 7sLRNPs. The 7sL-sc RNA is about ~300 ntd long and contains complementary sequences that lead to ds helical regions encompassing nearly 80% of its length. The scRNAs can be cleaved with micrococcal nuclease at 100th ntd, into two pieces, one has Alu domain and the other is S-domain. Both domains are associated with a set of proteins like p-9/p14, p-68/p-72, p-19 and p-54. The whole complex is called SRP particle (signal recognition protein), for it recognizes signal polypeptides of those proteins which are destined to be membrane or secretary proteins. The SRP complex is responsible for transferring the translating machinery and doc it on to docking proteins found on Endoplasmic reticulum. Similar to 7SL there are few more sc RNAs, whose function is not well understood.
Sn RNPS:
- Proteins bound to Sc and Sn RNAs are referred to as Sc-RNPs and Sn RNPS (Scyrps and Snurps respectively).
- Nucleus contains hundreds of such proteins that bind to Sn RNAs. The said proteins bind Sn RNAs to specific secondary structures with specific sequences.
- Some of the proteins are common to all Sn RNAs and they bind to a region called Sm domain. The said proteins contain RRNP motifs.
- The proteins are approximately 90 aa long which include some highly conserved RNA binding motifs; contain an 8 amino acid sequence towards C- terminal end and a hydrophobic aa sequence at N-terminal end. The Sn proteins have 4 beta strands and two alpha strands. It is believed that the RNAs bind to N-terminals of beta strands and C terminals of alpha strands. Many of these proteins have RNA binding motifs such as RGG box (Arginine-Glycine-Glycine), KH motif (lysine-Histidine), SR motif (Serine-Arginine) and similar structural features, which facilitate the proteins to bind and organize the sec structure of the SnRNAs.
- Some proteins are specific-to-specific Sn RNAs. At least 4-5 different proteins do associate with each snRNA species. Association of proteins with Sn or sc RNAs prevents unwanted secondary structural formation of newly synthesized Sn RNAs; facilitate protein-protein interactions, facilitate rRNA-RNA interactions and RNA-protein interactions.
- Each SnRNAs with SnRNPs act as individual units and interact with pre RNAs and bind to specific sequences or structural motifs. The binding leads to interaction among the snRNA-SnRNPs and form a complex. Such complexes bound to pre-mRNA modulate the mRNA to a variety of conformational changes and splicing reactions. In this process, individual snRNA-RNP act as dynamic functional units, where they interact with specific domains of pre-mRNA, bring about folding in pre-mRNAs, interact with other snRNAs and assemble into a large complex of units and ultimately bring about the most complex splicing reactions. The complexes are as big as 60s in size and they are called Spliceosomes.
Common snRNPs are those found bound to all snRNAs (U RNAs). They are also called core proteins.
Small common core group of proteins:
|
B‘=29KDa |
D3=18Kda |
|
B=16Kda |
E=13Kda |
|
D1=16Kda |
F=12Kda |
|
D2=16.5 |
G=11Kda |
|
|
|
SnRnps specific to Sn RNAs:
U1= A=34Kda, C=22Kda, 70Kda,
U2= 12 others,
U4= two other U$ and U6 specific proteins,
U5= 9 other proteins (25Kda),
All are identified using anti-U1-Rnp and anti sm Abs

Spliceosomal U snRNP components conserved inArabidopsis. Subunits encoded by two or more genes are highlighted in bold and characterized gene mutations are indicated; http://journal.frontiersin.org/ in red.
SnRNPs function:
- They assist assembly and formation of RNA secondary structure.
- Assist in transport,
- Assist in snRNA-SnRNA and snRNA-mRNA interactions,
- Facilitate the assembly of spliceosomes,
- They are involved in splicing process
Sn-RNA biogenesis:
Most of the sn RNAs are coded by specific genes spread over different chromosomes. See the table of snRNAs locations in Arabidopsis thaliana. Most of the Sn RNAs like U1, U2, U3, U4, U5, U6, U7, U11, U12 transcribed by RNA pol II and U6 is transcribed by RNA pol III also whose promoter elements are different.
U3 RNA is transcribed by RNA Pol II with 210 ntds, yet no 5’cap, but localized in the nucleolus and involved in processing pre rRNA. It contains two boxes A and B; where A cross links to rRNA and B interacts with rRNA. This is involved in the first cleavage at 5’ side of the precursor rRNA. The U6 RNA is transcribed by RNAP III, but it is involved in processing pre mRNAs. The U7 RNA is different from the others for it is involved in processing Histone mRNAs. It has a mono methyl cap but no poly-A tail.
Correctly regulated gene expression is important for healthy development and the maintenance of cellular homeostasis. Disruptions in transcriptional regulation are observed in many diseases including cancer. Moreover, viruses subvert the cellular transcriptional programs for their own benefit during viral infection.
Therefore, it is important to understand the transcriptional mechanisms that underlie control of gene expression. Human snRNA genes encode short stable RNAs that as part of specialized protein- SnRNA complexes typically are involved in processing other RNA molecules. Sn RNAs are essential for mRNA, rRNA processing.
In addition, other SnRNAs process RNA primers required for mitochondrial DNA replication. Thus, SnRNAs play pivotal roles in numerous cellular processes. The genes for these SnRNAs are unusual because they have similar promoter architectures and they are transcribed by RNA polymerase II while others are transcribed by RNA polymerase III. These differences in polymerase choice make this an interesting system for understanding mechanisms of transcription for both RNA polymerases II and III.

RNAP II promoter core elements; http://genesdev.cshlp.org/
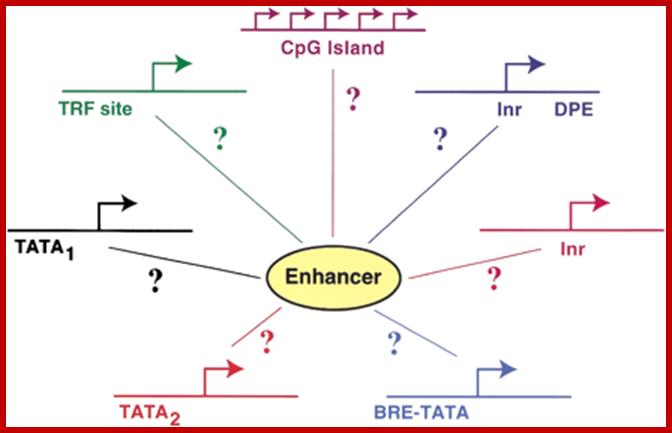
Canonical TATA box (∼TATAAA) or a noncanonical TATA element (i.e., an AT-rich region lacking a TATAAA-like motif).;Enhancer act with a variety of Core promoters; http://genesdev.cshlp.org/

The figure shows snRNA gene promoters and RNAPs that transcribe; note snU2 snRNA promoter transcribed by RNAPII contains PSE and DSE elements but no TATA box, on the contrary snRNA genes transcribed by RNAP III contains PSE, DSE and TATA box elements. Both require polymerase specific PTFs (promoter specific factors) for transcription. U6 RNA genes are transcribed by RNAP II and RNAPIII. Dr.S.Murphy; Molecular Biology; http://users.path.ox.ac.uk/

RNA Pol II associated components; http://alfa-img.com/
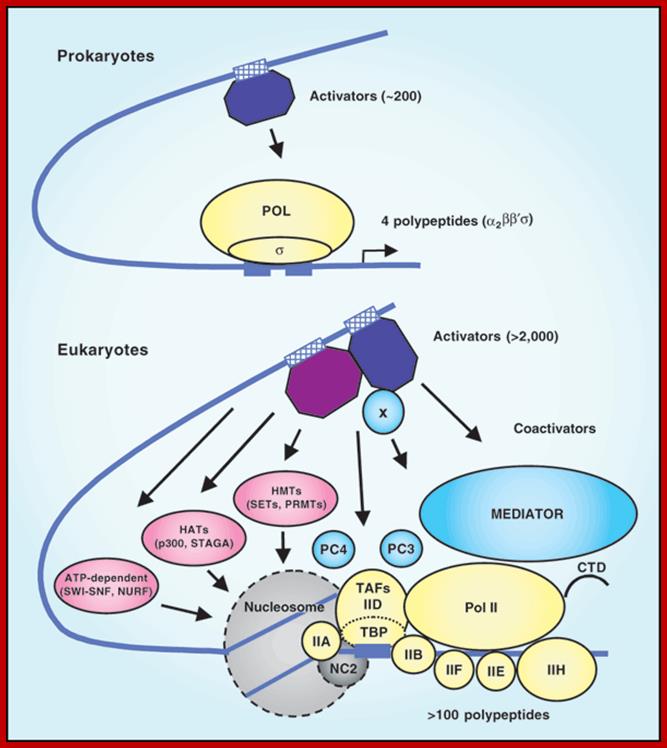
The eukaryotic transcriptional machinery: complexities and mechanisms unforeseen;
Robert G Roeder. http://www.nature.com/
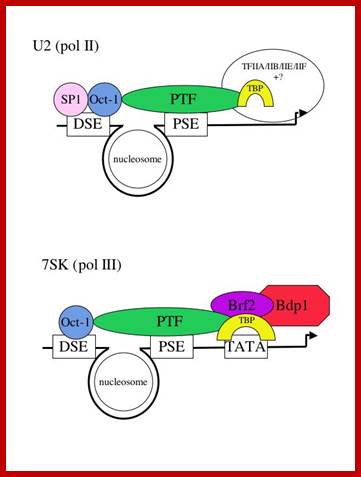
Structure of snRNA genes and known transcription factors; http://users.path.ox.ac.uk/
Several of the transcription factors involved in the expression of the human snRNA genes have already been identified and characterized and we can therefore more easily investigate their function. The promoter elements in a pol II- and a pol III-transcribed snRNA gene are shown below (Fig 1) with the cognate factors that have been identified so far. In vivo foot printing studies suggest that a nucleosome brings the DSE and PSE together.
The major difference between the pol II- and pol III-transcribed genes is the presence or absence of a TATA box-the rest of the elements are largely interchangeable (see refs. 2 and 3). Not all the factors required for transcription of these genes have been identified, as indicated by question marks. The pol II-transcribed snRNA genes share TBP/IIA/IIB/IIE and IIF with protein-coding genes that are also transcribed by pol II. The pol III-transcribed snRNA genes share components of the IIIB complex with tRNA genes that are also transcribed by pol III. In addition, these genes require an snRNA gene-specific factor-Brf2. Dr.S.Murphy; http://users.path.ox.ac.uk/

Linking transcription to 3' box-dependent RNA processing through the CTD of pol II. 1) SnRNA-specific 3' processing factor(s) interact with snRNA gene promoter-specific factors (e.g. PTF) before recruitment of pol II. 2)The processing factor(s) associate with the CTD after phosphorylation at initiation (by e.g. CDK7 or CDK9, see refs. 11 and 12) and travel with the polymerase until transcription of the 3' box. 3) RNA processing occurs after factors interact with the 3' box and the reaction is activated by the phospho-CTD. 4) Termination occurs after release of the RNA processing factors and dephosphorylation of the pol II CTD after which another transcription cycle can begin. 5) In the U1 snRNA gene termination of transcription is facilitated by a protein binding to a specific region located immediately downstream of the 3' box The snRNA transcripts are terminated and processed by specific factors, and they are associated with phosphorylated CTD tail; for termination 3’box, is an RNA processing element analogous to poly-A site, is required. Dr.S.Murphy; http://users.path.ox.ac.uk/
(1) How does SNAPc function in transcription by both RNA polymerase II and III?
(2) How does SNAPc communicate with other specialized factors that are dedicated for transcription by only RNA polymerase II or III?
(3) Is SNAPc the target of transcription regulatory proteins?
(4) Does SNAPc selectively mediate transcription of some snRNA genes depending upon the demands of the cell?
Answers to these questions are important for understanding the complicated network of transcriptional regulation in the cell.
Sno RNAs and Sno-RNPs:
Sno RNAs:
Another class of small molecular weight noncoding RNAs is small Mol.wt nucleolar RNAs for they are localized in the nucleolus, the loci where rRNA are transcribed and processed to be assembled into functional ribosomes. More than 90% of the RNA produced in the cell is rRNA and the nucleolus is the most active region all the time for it is in this locus rRNA is transcribed and processed and assembled using imported ribo proteins into ribosome structure; then the same transported into cytoplasm through nuclear membrane pore complex. All these noncoding RNAs found in the nucleolus that are involved in processing rRNA are called snoRNAs and their associated proteins are called snoRnps.
Nucleus also contains another very important structure involved in processing a variety of RNAs-called Cajal bodies. Eukaryotic ribosomal RNAs are transcribed as long precursors, they are sequence wise cut and trimmed, and then they are folded into 3-D structures in association with riboproteins. Before pre-rRNA is cut, ribosomal RNAs are modified at specific positions (in sequence specific manner) in the form of 2’O methylation of ribose sugars and conversion of Uridine into pseudo Uridine in sequence specific and site specific manner. These modifications help nucleases to cut and trim precursor rRNAs in specific manner. These modifications also help in binding of specific riboproteins to specific sites and folding into 3-D structure. They also help in ribosomal functions- binding of mRNAs, binding of tRNAs, binding of translational factors with GTP, performing peptide bond formation and movement of ribosomes along the polynucleotide mRNA.
Methylation and pseudouridinylation is performed by a special small molecular weight Sno RNAs and specific SnoRNPS. There are 100 or more known sno RNA genes. Most common snoRNAs are U26, U31, U48, U50, U73, U74, U80, and U81;
Drosophila contains 26 different sno RNA genes. Arabidopsis contains 25 sno RNA genes but in other plants there are 66 or more C/D sno RNA genes.
There are two classes of snoRNA; one class called C/D Snos and the second called H/ACA snoRNAs. First, specific class of enzymes add methyl groups to 2’O ribose sugar and the second class convert Uridines to psuedouridines. Methylation is performed SAM enzyme and Psu is performed by Pseudourine synthase.
C/D Snos: There are 40 different sno RNAs in vertebrates, 63 C/D Snos in drosophila; they are 60-300ntds long. Abundance 200,000 per cell; Best known sno RNAs are – U3, U4, U8, U22 and 7-2MRP RNAs.
C/D sno RNA contain two boxes one C-box and the other D-box with a central hexamer sequence.
5’ ---C= RUGAUGA-----XXXXXX----D = CUGA---3’; methylated at 5th ntds from D box
Associated proteins:
Fibrillarin proteins 65 and 68 KD, they contain methylase domain.
Nop-56KD,
Nop-58KD,
NOP-5,
Snu 13p/15.5 KD,
7-2 MRP RNA,
SnoRNA s that are transcribed by RNA Pol II are are added at 5’ with trimethylated cap; but those derived from Introns don’t have cap. In some plants sno RNAs are transcribed by RNA Pol III, they contain gamma methyl Cap. Most of the sno RNAs are trimmed at their 3’ end similar to Histone mRNA 3’ processing.
H/ACA Sno RNAs:
There are 20 snoRNA associated with specific proteins.
In drosophila there are 56H/ACA snoRNAs
In humans there are 95 sites for pseudouridinylation.
Yeast contain 43 sites and vertebrates contain >100 sites
Associated proteins-
GAR1p or Dyskerin proteins.
Cbf5p- is pseudo Uridine synthase,
Nhp2p,
Nop10p,
Pop1-3 protein,
MRP 7-2RNA.
Structurally H/ACA sno RNAs contain two stem loops; in between two loops it has ---ANANNA--- sequence called H region , at the 3’ end of the second loop it has ACANNN—3’ region. Each stem loops have two open regions. When RNA base pairs, two of the Us in the bottom open region remains unpaired and the right U is converted to pseudouridine.
Orphan Sno RNAs:
There are many sno RNA whose target is not known, so they are called Oprphan SnoRNAs.
Cajal bodies:
They are found in the nuclear sap outside the nucleolus. They contain both GAR-p and Fibrillarins proteins; they also contain C/D and H/ACS sno RNAs. They also contain coiled coil proteins called Coilins, so the name Cajal bodies. The structure is also loaded with RNAP I, II and III polymerases, very many nuclear transcripts, transcription factors, SR proteins Poly-A adenylation factors snurposomes and many RNA processing enzymes.
U6 RNA was first found to be modified by enzymes found in Cajal bodies. U6 sn RNA does contain cap and it is transcribed by RNA Pol III and also by RNAPII. U6 RNAs contain 3 psuedouridines, eight 2’O methyl sites.
Sca-RNAs- Cajal bodies were found to process U1, U2, U4, U5 and U6 sn RNAs. They also contain Sno RNAs such as U85, which contain both C/D and H/ACA domains, so they can perform both methylation and pseudouridinylation of sn RNAs such as U1, U2, U4, U5 and U6, totally they contain 30 2’Omethylation and 24 pseudouridinylation sites. The Sno RNAs found are called Sca RNAs. Cajal bodies also modify several tRNAs and even some mRNAs
Biogenesis of Sno RNAs:
They are coded for by genomic DNA, where they are derived either from RNA Pol-II transcribed gene as monocistronic mode or polycistronic mode. Such transcripts also undergo processing; those transcribed by RNAP II may contain Cap structure at 5’end, however their 3’ end is processed for they don’t have poly-A tail. Many of such Sno RNA genes are found as tandem repeats. There is another class of Sno RNAs which are derived from Introns of pre-mRNAs. The intron is released after cis splicing. The released structure looks like a lariat. This structure is debranched and processed to functional Sno RNA.

Genomic organization of snoRNA coding units. Schematic representation of the different types of genomic location of snoRNA genes. The snoRNA coding units endowed with independent promoters (top) and those located within introns (middle) are transcribed by RNA polymerase II. Frequently, neighbouring introns of the same host gene contains snoRNA coding units with a one-gene-per-intron distribution. In such cases, the snoRNA coding units have been considered as “intronic individual” (Table 1), even though several different snoRNAs can originate from the same precursor transcript.;Giorgio Diec et al;http://www.sciencedirect.com/

Sno RNA genes; http://mcmanuslab.ucsf.edu/

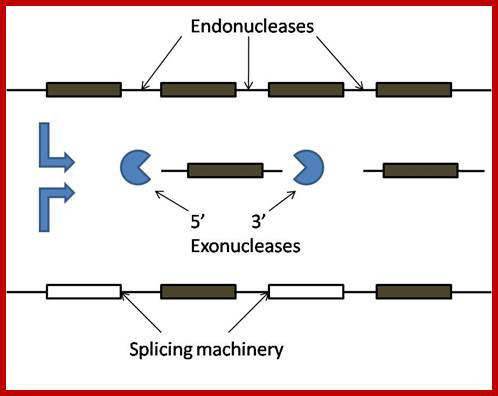
Sno RNA genes-mono and polycistronic and processing the transcripts first by endonucleases; http://mcmanuslab.ucsf.edu/
RNA Processing:
Some snoRNAs are involved in the processing of pre-rRNAs rather than nucleotide modification. These include C/D RNAs U3, U8, U14, and U22, as well as H/ACA RNAs snR10, snR30, E2 and E3. These snoRNAs direct processing machinery to specific cleavage sites on pre-rRNAs (Venema et al. 1999; Kressler et al. 1999). http://mcmanuslab.ucsf.edu/
Gene structure and transcription:
Eukaryotic cells exhibit differences in both the genetic organization and mechanisms of biosynthesis of snoRNAs. While only a few vertebrate snoRNAs involved in rRNA processing (not modification), such as U3 or U8 are trancsribed as independent genes with their own promoter nearly all snoRNAs identified in yeast are generated in this manner. On the other hand, plants often generate polycistronic transcripts from which multiple snoRNAs are processed. In vertebrates, all of the snoRNAs that are known to guide nucleotide modifications are located within introns of genes transcribed via RNA polyermase II (See Figure). Many snoRNAs are embedded within introns of genes that themselves are involved in ribosome biogenesis (Terms et al. 2002, Steitz et al 1999). http://mcmanuslab.ucsf.edu/
snoRNA processing: sn RNA transcripts are produced by their specific snoRNA genes (either monocistronic and polycistronic) or they are derived from introns’ First they are cleaved by endonucleases such as Rnt1p, which cleaves at stem loop structure. Once thery are released they are processed by exonucleases from 5’ end 3’ end. Then they are associated with their snoRNPs to perform functions in rRNA processing. Important snoRNA and their snoRNPS are C/D and H/ACA sno RNA-SNRPs.
RNAi (RNA interference):
RNA interference is a novel system by which specific mRNAs are identified and block translation and possibly degradation of the mRNA. There are two kinds of small mRNA interfering RNAs called siRNAs (small interference RNAs) and Mi RNA (small mRNA interference RNAs). Can these be included in what is called noncoding RNAs?! Yes. In recent times the presence noncoding RNAs is gaining recognition and they are different from RNAi populations. What is their function is yet to be discerned though there are speculations about them.
SiRNAs:
SiRNAs act as silencers of mRNAs , silencers of viral RNAs and even the transcripts derived from retrospons and transposons, however it is to be noted the slicer activity is specific; thus they regulate gene expression at translation level or what is called post transcription level. In 1998, in an unsuspected situation, researchers found that a dsRNA found to be responsible for the degradation of a specific mRNA. Though this was established as a fact in plants in later years, it was suspected that siRNAs act as viral RNA mediated cross protection molecules. This was by the production of dsRNA, and by the action of Dicer and Risc, leads to the degradation of viral genomic RNA. In animal system any dsRNA produced can trigger INF response which acts through kinase eIF2a.

Primery miRNA transcript is stem loop structure (Sh) and the same processed by Drosha then it is exported and further it is processed by Dicer complex. The mi RNA 22 nts long finds its target and binds, this leads to inactivation of the mRNA or degradation of it. MiR 143 and miR-145;Advaces in genetics; Ashraf Yusuf Rangrez et al,

Small Interefering RNA; Diagram illustrates the major steps in biogenesis and subsequence siRNA mediated silencing; http://mcmanuslab.ucsf.edu/
The cascade leading to the generation of mature siRNA begins with transcription by RNA polymerase II (in animals), RNA polymerase III (from a shRNA template), or RNA polymerase IV (in plants), forming double stranded RNA (dsRNA). These dsRNA are recognized by the RNA binding domain (RBD) of the Dicer complex, which contains a pair of RNaseIII type endonuclease catalytic domains. The dimeric Dicer complex then cleaves the dsRNA into ~21-28 nucleotide siRNA duplexes containing 2-nucleotide 3' overhangs with 5' phosphate and 3' hydroxyl termini, which are bound by the Dicer PAZ (Piwi Argonaute Zwille) domain.
Production of Transgenic tomato plants against ToLCV using si/mi RNA teachnique-grkraj,B,lore
In fact many transgenic plants were produced resistant to RNA viral infection. Transgenic plants were made by converting viral RNA into cDNA and truncated it to have only capsid protein segment without any ORF sequence another with ORF; then they were cloned into plant vectors end to end in apposite orientation to be expressed under CaMV 35S promoter. Similarly ToLCV viral DNA was cloned into CaMV 35S promoter one with complete capsid protein segment and another with a fragment lack of ORF. The transcript produced in one case produced proteins and the other could not translate for it had no ORF, but the transcript with no ORF somehow generated partial ds RNA structure. This dsRNA was found in cells infected with ToMV or ToLCV. The ds RNA is processed in to 23 bp long short fragments by DICERs or Dicers. Then such fragments are loaded with RISC components. Once loaded with RISC proteins they cleave one of the strands leaving one strand intact. The single stranded RNA base pairs are presented to viral RNAs. Wherever, they have complementary base pairing. Risc proteins endowed with RNases are activated, which degrades the base paired mRNAs; RNA Induced Silencing Complex (RISC) is responsible for this. RISC consists of many Argonaut and AGO related family of proteins, some of which are responsible for the degradation of base paired RNAs. This is one of the modes for developing resistance against plant viral infections.
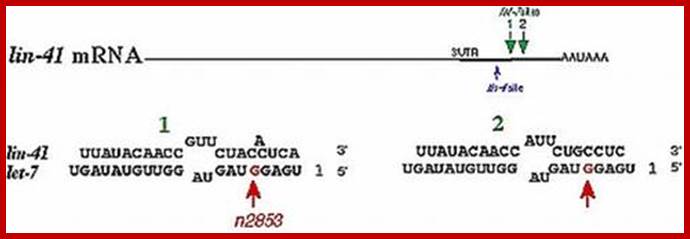
RNAi mechanism was found in Coenorhabdites elegans a worm acting on Lin41 mRNA during developmental stages.
SiRNAs are found in all systems including plants, animals and yeasts. siRNA are derived from long dsRNAs or they are derived from viral transcripts or by many of the transposons and retrospons; the transcripts because of complimentary base sequences they base pair, to generate a double stranded RNA of 80 to 90bp. Such dsRNA transcripts are recognized by a protein complex called Dicer.
These Dicers are similar to RNase III. They bind to ds RNAs and cleave to generate 21-23 base pair long RNA molecules called SiRNA. These have characteristic 2 ntds overhangs at 3’ ends. These molecules interact with genes or gene transcripts and inhibit their expression in three ways. First, they base pair with mRNAs, which have complementary, sequences and prevent their translation, second if the base pairing is not complete but very partial, and then they induce enzyme, which degrade mRNA.
The third method is they bind to specific proteins and get transported into the nucleus, where they interact with the DNA that has homologous sequence and inhibit the expression of that gene.

http://sgugenetics.pbworks.com/

rmor@sgu.edu; http://sgugenetics.pbworks.com/
Small interfering RNA is typically found to be a small piece of double stranded RNA, about 20-25 nucleotides long, with 2 nucleotides (3’) overhanging at each end. A 5’ phosphate group and 3’ hydroxyl (OH) group exists on each strand. siRNAs are products of long dsRN-As or small hairpin RNAs. Small hairpin RNAs, are single strands of RNA which fold s back and base-pairs with itself; on one end there is a little loop and the other there is a single- stranded tail.
The silencing mechanism uses a protein complex called Risc i.e. RNA induced silencing complex. The Risc contains SiRNAs and several proteins. Some of the proteins found in RiSc are found to be members of Argonaut family, which are believed to interact with RNA components. When the Risc complex binds to such ds-SiRNA, it opens up the dsRNA into ssRNA strands in ATP dependent manner; it is the ssRNA that activates the RiSc complex. Once it is activated it facilitates the Si-ssRNA to base pair with mRNAs which have complementary base sequences. When the siRNA base pairs with its homologous mRNAs, another set of enzymes become active which recognize such SiRNA-mRNA complex and degrade them.
In Saccharomyces pombe, Si RNAs develop from the transcripts generated by the CEN-DNA. The pre-SiRNA is in the form of looped ds RNA, which on association with Dicer; it is cleaved to generate Si RNA, which target a specific transcript of the chromosomal DNA to silence the expression of it.
This mechanism of gene silencing can be used to silence a gene. It is possible to generate a transgenic animal or plant with a specific SiRNA-gene (constructed specially for this purpose) against a specific host gene, thus this technology can be employed in therapy or to find out the effect of silencing a gene on overall morphology and function of an organism. A cloned gene for generating a SiRNA was introduced into bacterial cells. Worms are fed with such gene containing bacteria, where bacteria express dsRNA, which when enter into host cells or germ line cells, they affect the cell phenotype. Using this technology 86% of the worm genes has been knocked out to see what is the effect of such gene knockouts on the organism.
Different classes of Si RNAs:
Transcription initiation RNA (tiRNA): Tiny, or tiRNAs, were discovered by Next Generation Sequencing (NGS) studies. RNA libraries were prepared from specific size fractions of capped messages. The resulting libraries were sequenced on the Roche FLX Genome Sequencing system and the data were aligned to human genome build 36.1 and compared to transcription start sites (TSS) defined by Ref Gene (NCBI). The authors reasoned the previous deep-sequencing studies missed these RNA molecules because they tend to be disregarded as low-abundance spurious, or degradation products. However, because they can be cloned, they must have a 5’ phosphate and, when aligned to genomic sequences, the NGS reads cluster in a non-random fashion around TSSs.
Trans-acting short interfering RNAs (tasiRNA): They are ~21 nt long small RNAs that require endogenous transcript as template that are converted to dsRNA by RNA dependent RNA polymerase (RdRP) activity and subsequently requires the downstream activity of DCL4 and AGO7 to generate functional tasiRNAs. Animals like humans, flies etc., which lack RdRP, are devoid of these small RNA species. Tasi-RNAs resemble miRNAs both in size and function and are involved in targeting non-identical mRNAs. It has been demonstrated that miRNA primed transcripts recruit RdRP that consequently generate tasiRNAs, thereby setting an example of small RNAs mediated regulation of other small RNAs. For instance, miR390 binds to and induces the RdRP activity on primary transcripts and convert them to long dsRNA. In Arabidopsis, the six tasiRNA genes are present that target Auxin Response Factors (ARFs) and MYB transcription factor. One of the recently identified tasiRNA locus, TAS4, has been demonstrated to generate siRNA that targets the transcript at a site which is different from the miR828 cleavage site. This indicates towards the possibility of parallel evolution of tasiRNA, miRNA and their common target in plants.
In an alternate pathway, RdRP can also act on aberrant transcripts (usually viral transcripts) converting them to dsRNA and this mechanism is likely to be responsible in preventing cell from any erroneous transcription event that might affect cellular integrity.
(ii) Repeat-associated short interfering RNAs (rasi-RNAs): They are ~24-26 nt long products of DCL3 activity on dsRNAs formed during unchecked transcription event, usually retro-transposons loci. These loci are generally methylated which prevent transcription through such regions. Like tasiRNA, these also require RdRP for amplifying small RNA pool. Rasi-RNAs play important role(s) during gametogenesis in flies, worms and mammals by modulating the chromatin status, and silencing viral transcripts by recruiting histone modifying proteins (Figure 1, step h).
(iii) Scan RNA (scn RNA): These are another type of relatively long (~29 nts) siRNAs have been reported from protozoan Tetrahymena thermophila. This organism exhibits nuclear dimorphism differing by ~15% at the sequence level. During conjugation, scn RNAs derived from micro-nucleus are generated (reproductive nuclei) and eliminate corresponding loci from its own genome while giving birth to macro-nucleus. This phenomenon requires Argonaute like Twi1 protein, and seems to be an ultimate form of RNA interference wherein organism can efficiently utilize small RNA to produce modified versions of genome from the existing ones.
(iv) Long siRNAs (lsiRNAs): They are recently introduced class of siRNAs that are 30-40 nt in length and are induced in response to bacterial infection or growth conditions. Discovered in Arabidopsis, the generation of lsiRNAs require DCL1, DCL4 and AGO7 proteins and depend on other established members of both siRNA and miRNA pathway e.g. RDR6, HYL1, HEN1 etc. One of the lsiRNAs targets a protein that confers resistance against bacterial infection. Interestingly, these lsiRNAs unlike other siRNAs are believed to mediate target degradation by a mechanism previously known in animals but not in plants.
miRNAs:
Another class of RNAi’s similar to that of siRNAs. They also silence or repress a specific gene expression. Most of the miRNAs are 21-24 nucleotide long transcripts; actually they derive from a longer transcripts (70-90 ntds), which are double stranded with a closed loop on one side. These get associated with Dicer proteins or Dicer-like proteins. The Dicers on binding to ds-looped transcripts unwind ds RNAs and its RNase cleaves the ds-transcript into 22 ntds long transcripts. The dsRNA contain some imperfect base pairing unlike siRNAs. These miRNAs are loaded with Risc protein via Risc loading components. Once the Risc proteins are bound, the ds RNA unwinded to single strand form and the asymmetric strand is used for base pairing with specific mRNA at 3’ region. Thus the target mRNA is prevented from translation.
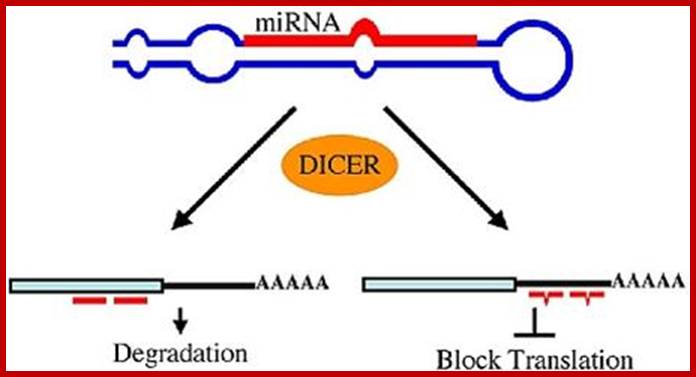
miRNAs use two mechanisms to exert gene regulation. Some animal miRNAs can bind to mRNA targets with exact complementarity and induce the RNAi pathway. miRNAs also bind to targets with imperfect complementarity and block translation. There is no evidence that C. elegans miRNAs use the former pathway.
Monica C. Vella, Frank J. Slack; http://www.wormbook.org/
For the first time these miRNAs were observed in Caenorhabditis elegans. During the development of the worm these are produced in stage specific manner and they knock out specific gene expression through destroying its mRNA.

Linda Lin; http://www.scilogs.com/
Such genes are found in worms (~55-120 or more), human beings (~300) and other animals and also plants (very large numbers). This kind of genetic operation of suppression of specific gene products at a particular stage of development is a par excellent example of differential gene expression during development.
![]()
![A closer look at the model for siRNA guide-strand tethering by AGO2 and target-mRNA recognition and slicing. The terminal 5’ monophosphate group of the guide strand tucks in between the MID and PIWI domains of AGO2. Meanwhile, AGO2’s PAZ domain has a hydrophobic pocket that specifically recognizes the guide-strands 3’ dinucleotide overhang. This positioning opens up siRNA guide nucleotides 2-8, the “seed region,” for base pairing with complementary target mRNA, and next base pairing at nucleotides 10-11 correctly orients the scissile phosphate between these two for cleavage by AGO2’s PIWI domain, which houses the protein’s “slicer” activity [12].](Ribose_Nucleic_Acid4-Small_Molecular_Weight_RNAs_files/image039.jpg)

Use of RNAi in medical use as atherapy is very promising, but it delivery and its stability is still aproblem.Therapeutic siRNA: Principles, Challenges, and Strategies; Gavrilov K, Saltzman WM - Yale J Biol Med (2012); http://openi.nlm.nih.gov/
In the case of C. elegans, the gene Lin 4 regulates the expression of Lin14 gene. Lin4 transcript is 22 ntds long. The gene is transcribed by RNA pol-II, which produces a non-coding transcript; it means it cannot generate any proteins for it is lacking ORF. The transcript has a ten base sequence, which is also found in Lin 14 gene, but it is repeated seven times at the 3’end of the gene. The Lin-4 transcript base pairs with Lin-14 transcript and inhibits its translation or the Lin-14 mRNA is destroyed by an RNase-III associated with Risc complex. It is interesting to note that the 3’ end of the mRNA is used for the control of its expression. Many of the miRNA in the worm are contained in a large complex riboproteins of 15s. Many of the miRNAs of the worms have complementary sequences in human cells and also in plants. Among a large but diverse number of miRNA found in plants, 16 of miRNAs are also conserved and found in Rice plants, which suggests a common origin of regulatory system.
The above mechanisms are related to RNAi mediated gene silencing or post-transcriptional gene silencing mechanisms. Infection of plant cells with RNA viruses, leads to the formation of dsRNA, which suppresses plant cell RNAs from plant genome, this has drastic effect. It is also assumed that the signals in the form of dsRNA can to spread to other region of the plant body as viruses do, crossing cell boundaries through plasmodesmata (cytoplasmic bridges found across the common wall found between two cells. Furthermore the signals can be amplified by some novel polymerases that use these SiRNAs as templates.
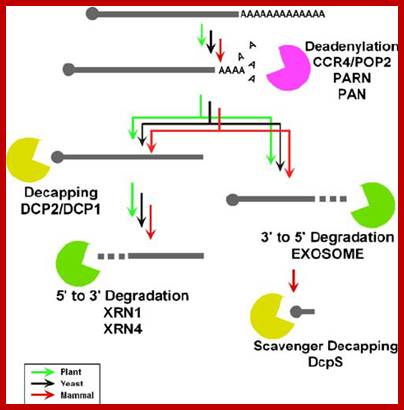
Control
of gene expression is exerted by multiple steps such as transcription, mRNA
processing, mRNA export, mRNA degradation, translation, and posttranslational
events. Recent discovery of small RNAs has enhanced the impact of
posttranscriptional regulation, in particular, alterations in mRNA stability in
the regulation of gene expression. Therefore, mRNA turnover is an important
process not only for setting the basal level of gene expression but also as a
regulatory step. Compared to the mechanism of transcription, much less
information is available regarding mRNA degradation machineries. However, in
the past several years, various components involved in the mRNA degradation
process have been identified in eukaryotes. In particular, progress in the
plant field has revealed the involvement of mRNA turnover in a wide variety of
developmental processes and hormonal responses. Here, we provide an overview of
machineries involved in general mRNA degradation and mRNA surveillance systems
in plants.
mRNA Degradation Machinery in Plants (PDF Download Available).
Available from: Yukako Chiba https://www.researchgate.net/publication/225787089_mRNA_Degradation_Machinery_in_Plants
[accessed Jun 10, 2017].
miRNA biogenesis and mechanisms of action:
MicroRNA control of signal transduction: Masafumi Inui, Graziano Martello & Stefano Piccolo.
MicroRNAs (miRNAs) are transcribed as primary transcripts (pri-miRNAs) by RNA polymerase II. Each pri-miRNA contains one or more hairpin structures that are recognized and processed by the microprocessor complex, which consists of the RNase III type endonuclease Drosha and its partner, DGCR8 (see the figure). The microprocessor complex generates a 70-nucleotide stem loop known as the precursor miRNA (pre-miRNA), which is actively exported to the cytoplasm by exportin 5.
In the cytoplasm, the pre-miRNA is recognized by Dicer, another RNase III type endonuclease, and TAR RNA-binding protein (TRBP; also known as TARBP2). Dicer cleaves this precursor, generating a 20-nucleotide mature miRNA duplex. Generally, only one strand is selected as the biologically active mature miRNA and the other strand is degraded. The mature miRNA is loaded into the RNA-induced silencing complex (RISC), which contains Argonaute (Ago) proteins and the single-stranded miRNA. Mature miRNA allows the RISC to recognize target mRNAs through partial sequence complementarity with its target. In particular, perfect base pairing between the seed sequence of the miRNA (from the second to the eighth nucleotide) and the seed match sequences in the mRNA 3′ UTR are crucial. The RISC can inhibit the expression of the target mRNA through two main mechanisms that have several variations: removal of the polyA tail (deadenylation) by fostering the activity of deadenylases (such as CCR4–NOT), followed by mRNA degradation; and blockade of translation at the initiation step or at the elongation step; for example, by inhibiting eukaryotic initiation factor 4E (EIF4E) or causing ribosome stalling RISC-bound mRNA can be localized to sub-cytoplasmatic compartments, known as P-bodies, where they are reversibly stored or degraded.
Figure is modified, with permission, from Ref. 104Nature Reviews Genetics © 2008 Macmillan Publishers Ltd; all rights reserved. m7G, 7-methylguanosine cap; ORF, open reading frame.

http://www.gurdon.cam.ac.uk/~miskalab/research.php:http://www.gene-quantification.de/
A list of RNAis’ and their features
|
|
siRNAs |
miRNAs |
piRNAs |
21U-RNAs |
|
Length (in nts) |
21-24 |
~22 |
25-31 |
21 |
|
Requirement of dsRNA precursor |
Yes |
Yes |
No |
No |
|
Genomic location |
Dispersed throughout |
Dispersed throughout |
Discrete loci |
Chromosome IV |
|
Frequency (in %) of 5′ U Monophophate |
~80% |
~76% |
~94% |
100% |
|
Location of Biogenesis |
Cytoplasm/Nucleus |
Nucleus and Cytoplasm |
Nucleus? |
Nucleus |
|
Nature of gene |
Autonomous /clustered |
Autonomous |
Tightly clustered |
Autonomous |
|
Proteins strictly associated with biogenesis (animals) |
Dcr 2, AGO2 |
Dcr 1, AGO1, Drosha/ Pasha, Exportin-5 |
Piwi/ Aubergine, AGO3 |
? |
|
Detected in |
All eukaryotes studied |
All eukaryotes but S. cerevisiae |
Worms, Zebrafish mammals |
C. elegans, C. briggsae |
|
Expression |
All tissues |
Every tissue but few shows tissue specificity |
Male germ line cells |
All tissues |
|
Downstream effects |
Target cleavage, Chromatin remodeling, Translation repression, Genome reorganization |
Translation repression, Target cleavage, Chromatin remodeling? |
Genome organization, Enhances translation and mRNA stability |
Nucleosome phasing |
|
3´ end modification |
Yes |
Yes |
Yes |
Yes |
|
Mode of transcription |
Divergent but partial overlapping Convergent |
Autonomous |
Divergent |
Autonomous |
|
Strand biasedness |
Yes |
Yes |
High |
Yes |
|
Selection pressure |
No |
High |
No |
No |
|
Nature of transcript |
Polycistronic |
Polycistronic/ Monocistronic |
Polycistronic |
Monocistronic |
|
Potential tool without adverse effects |
Yes |
In plants |
No |
No |
Comparison between siRNAs and miRNAs among plants and animals
|
|
Animals |
Plants |
|
siRNAs |
Usually, single dicer involved in all types of siRNA generation |
Different dicers required in Arabidopsis (4), Rice (10) |
|
Redundancy at functional level not observed |
Major proteins (dicer, Argonaute) are functionally redundant |
|
|
Systemic spread requires SID-1 protein |
Systemic spread requires PRSP1 (cucurbits) and SNF2 (Arabidopsis) protein |
|
|
Target cleavage, DNA methylation |
Target cleavage, DNA methylation |
|
|
siRNAs can participate in genomic DNA elimination |
No such role attributed here |
|
|
miRNAs |
Generally, target repression |
Generally, target cleavage |
|
More than one miR can reside on pri-miR |
Strictly one miR from one pri-miR |
|
|
Target various mRNAs |
More biased towards TF transcripts |
|
|
Multiple miRNA binding sites per target |
Usually single with one exception |
|
|
More than one miRNA can bind target |
No report |
|
|
Duplex miRs are formed in cytosol |
Duplexes miRs are formed inside nucleus |
|
|
Mature miRNAs can be trafficked back to nucleus |
No such validated report |
|
|
Pri- or pre-miR are subjected to editing |
No such phenomena observed |
|
|
Repressed mRNAs are stored in special organelles called P-bodies |
No such structure observed |
|
|
|
|
|
Mirtrons: Mirtrons a novel system an emerging concept of microRNA biogenesis:
In addition to the canonical miRNA pathway, animals have been shown to follow yet another mode of miRNA biogenesis where intron sequences can produce miRNAs. The hypothesis had emerged through analyses derived from pyro-sequencing of the small RNA pool from Drosophila S2 cell lines where some miR and miR* reads were mapped to the intronic regions. Such miRNAs originating from introns were termed Mirtrons. Fourteen mirtrons from Drosophila and four mirtrons from C. elegans have been identified so far. Given that both protein-coding and non-coding genes possess introns, it is predicted that >80% of miRNA are derived from such sites.
Introns denote the region of the transcript, generally flanked by few conserved nucleotide residues, which are removed during RNA processing. The conserved GU-AG along with other sequences brings multiple proteins mediating the removal of the intron from the transcript. The characteristic 2′-5′ phosphodiester bond formed within the intron during splicing result in the formation of lariat like structures. Such structures are acted upon by lariat de-branching enzyme to release the single stranded RNA that is consequently degraded by various nucleases present. However, intron sequences having potential to form hairpin like structures might recruit proteins of miRNA pathway viz., Dcr1.
Mirtron generation deviates from the canonical miRNA pathway mainly in the non-requirement of DROSHA/DGCR8 proteins that remove the sequences flanking stem region of pri-miR (Figure 4). The unavailability of such sequences leads mirtronic pri-miRs to bypass the DROSHA step. This finding is contrary to the previous observation where Drosha was shown to act on intron prior to its splicing [101]. The knock down experiments with different transcripts involved in canonical miRNA biogenesis viz. Dicer1, Loquacious and Argonaute1, clearly supports that mirtron pathway do not require these proteins for their precursor generation. Moreover, the observation that Dcr-2 and RDR-2 (proteins participating in siRNA generation) knock down do not influence the expression of mirtrons, further ruled out any connection existing between the mirtron and siRNA biogenesis. The expression studies demonstrated that such introns can independently give rise to mature miRNAs, as is the case with miRNA genes. Moreover, like miRNAs, mirtrons also require AGO1 for maturation and follow cell-type specific expression
RNA silencing:
In higher systems like mammals and plants, ds RNA elicit a different type of response. The dsRNA, longer than 25 base pairs, activates an enzyme called RNA dependent protein kinase (PKR). The activated PKR phosphorylate eIF2a, which is a translation initiation factor; thus it is rendered inactive (similar to interferon mediated silencing eIF2) it also activates 2’-5’ oligo-Adenylate synthetase enzyme, which produces poly 2’-5’ Adenylate oligos. These oligos in turn activate RNase-L, which degrades all mRNAs. On the contrary if a shorter ds RNA of 21-13 base pairs long is introduced, it invokes specific degradation of complementary RNAs just like RNAi processes. In plants another phenomenon that was observed is that, when a plant gene is introduced into plants, the transgene often produces both sense and antisense transcripts. These transcripts act on specific plant gene transcripts and leading either to destruction of the mRNA or preventing it from translation. This phenomenon is referred to as Co suppression. In many cases dsRNAs can induce methylation of corresponding chromosomal DNA. Methylation, in Eukaryotes, is known to cause gene silencing.
A list of si/mi RNA associated proteins
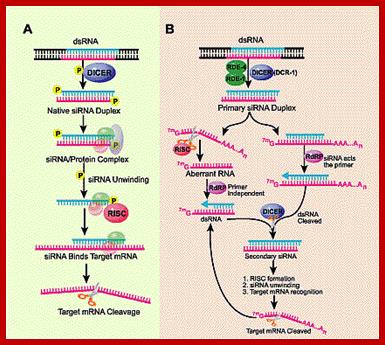
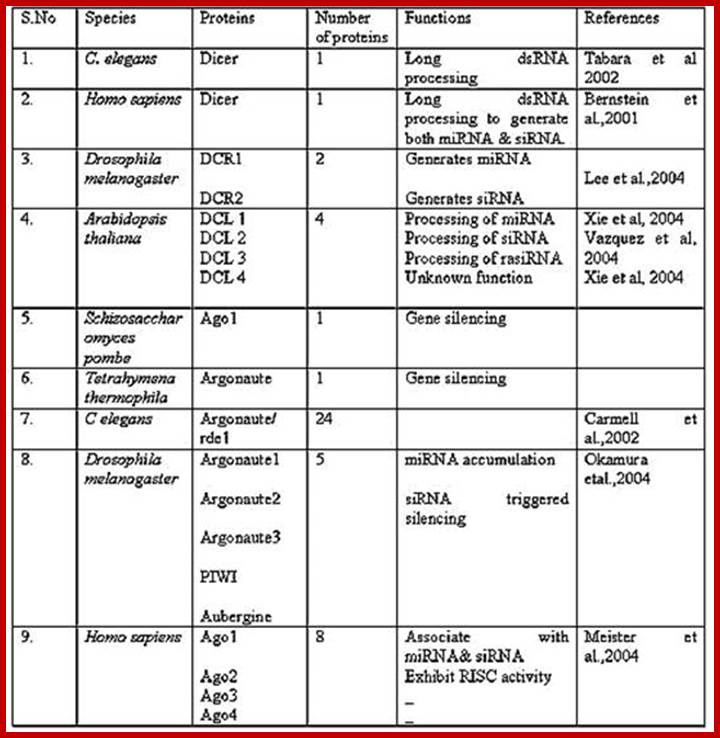
Mechanisms of post-transcriptional regulation by microRNAs: are the answers in sight?: Witold Filipowicz, Suvendra N. Bhattacharyya & Nahum Sonenberg.
Capped reporters containing multiple mRNA binding sites:
mRNAs containing a non-functional ApppN cap (instead of the 7-methylguanosine (m7G) cap) can be prepared by in vitro transcription with T7 phage RNA polymerase and either introduced into cells by transfection or used in studies in cell-free extracts. b | Mono-cistronic and bi-cistronic reporters containing a viral internal ribosomal entry sites (IRES). Reporters containing ApppN or pppN at the 5' end can be prepared by in vitro transcription and then transfected into cells. c | Reporters used to study the effects of tethering to mRNA of Argonaute (AGO) proteins or GW182 on protein synthesis.
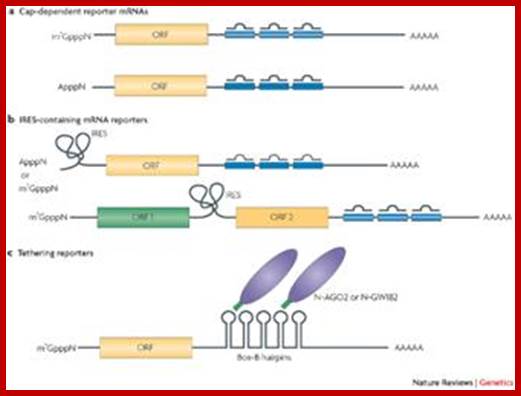
Nahum Sonenberg
The
investigated proteins are expressed as fusions with a phage ![]() N-peptide, which can bind the short Box-B hairpins that are
inserted to the mRNA 3' UTR. The
N-peptide, which can bind the short Box-B hairpins that are
inserted to the mRNA 3' UTR. The ![]() N-peptide–Box-B system can also be used to tether initiation
factors eIF4E or eIF4G to the intercistronic region of the bi-cistronic
reporter. Reporters that are generated in vitro and used
for either RNA transfection experiments or studies in cell-free extracts can be
prepared with or without the poly (A) tail. Reporters can also differ in the
number of miRNA binding sites that are present in the 3' UTR. Reporters that
are devoid of miRNA binding sites or that contain mutated sites used as
control.
N-peptide–Box-B system can also be used to tether initiation
factors eIF4E or eIF4G to the intercistronic region of the bi-cistronic
reporter. Reporters that are generated in vitro and used
for either RNA transfection experiments or studies in cell-free extracts can be
prepared with or without the poly (A) tail. Reporters can also differ in the
number of miRNA binding sites that are present in the 3' UTR. Reporters that
are devoid of miRNA binding sites or that contain mutated sites used as
control.
sRNAs:
In bacteria and few other systems there are a number of small RNAs, but non-coding, which are smaller in size and regulate the expression of certain genes at transcript level or translational level; they are called sRNAs. Such regulation has been found to operate in bacterial cells, which contain not less than 17 such sRNAs. Their expression and activity is mostly in response to certain conditions, can be stress or other similar types. These smaller RNA molecules themselves may have secondary structures, and they base pair to mRNAs wherever they find complementary sequences, thereby they inhibit translation. Base pairing at one position can change the structure of the RNA at other regions and make it non functional. These small molecules by base pairing may lead to the degradation of partner RNA.
The genes oxy-R and oxy-S exemplify the most interesting regulatory bacterial genes. These genes respond to oxidative stress. When the cells exposed to H2O2 or cells generate H2O2 due to certain chemical reactions, bacterial cells respond in activating oxy-R genes, which in turn activates Oxy-S genes. These sRNAs base pair with Flh mRNA and inhibit its translation. Another, but very important target is the transcript of Rpo gene. Rpo gene responds to stress signals and produces alternate sigma factors, which switch on different sets of genes in response to changed conditions. But in this case they inhibit its expression by base pairing to Rpo transcripts and prevent the cascade gene expressions, unnecessary for responding to oxidative stress. Besides oxy-R and oxy-S there other genes products such as Rpr-A and Dsr-A are also involved in such activities, thus they can be considered as global regulators.
Guide RNAs:
Some mitochondrial genomes mutated produce mRNA for cytochrome oxidase, which on translation does not produce the functional proteins. This is because the mRNA produced either contain some excess irrelevant nucleotides like Us or some Us missing or other changes in nucleotide sequences. To make such mRNA to contain correct coded information, such nucleotides have to be added or deleted to produce correct and meaningful mRNA.
To perform such correction mitochondria perse also produce some RNAs called Guide RNAs: The RNAs have sequences that can be used for correction of such mRNAs with defects. The guide RNA are used for base pairing with such defective RNAs and find what nucleotide is missing or extra, then enzymes associated with such guide RNA called editosomes, correct such defects by splicing the extra nucleotide or cleaving and adding the required nucleotide.
SRP RNAs: These RNAs are transcribed by RNAP III; these often called 7s RNAs or ScRNAs (small molecular weight cytoplasmic RNAs). In bacteria and chloroplasts one finds a similar RNA called 4.5sRNA (Ffh-ftsy RNA) which is involved in protein transport. These are involved in docking the ribosomes translating destined to membrane and such targets. From the free translating system, using specific signal sequences they bind to such nascent emerging polypeptide N-terminal region and recognize such sequences and transport the whole complex on to endoplasmic reticulum in what is called co-translation process. In this process the sc RNA with its fine tuned secondary structures get associated with specific proteins to form what is called SRP complex. This RNA is 320 350 ntds long and has very specific secondary structural features.
RNA-protein (ribonucleoprotein) complexes, such as the ribosome, signal recognition particle (SRP), spliceosome, and telomerase, carry out essential functions inside cells. We are currently investigating the structural mechanism of SRP-mediated co-translational translocation of proteins across or into cell membranes. In this vital cellular process, SRP recognizes the hydrophobic signal sequence of the nascent polypeptide emerging from the ribosome, resulting in transient elongation arrest in eukaryotes, and targets the ribosome to the membrane via a GTP-dependent interaction with the SRP receptor (SR).
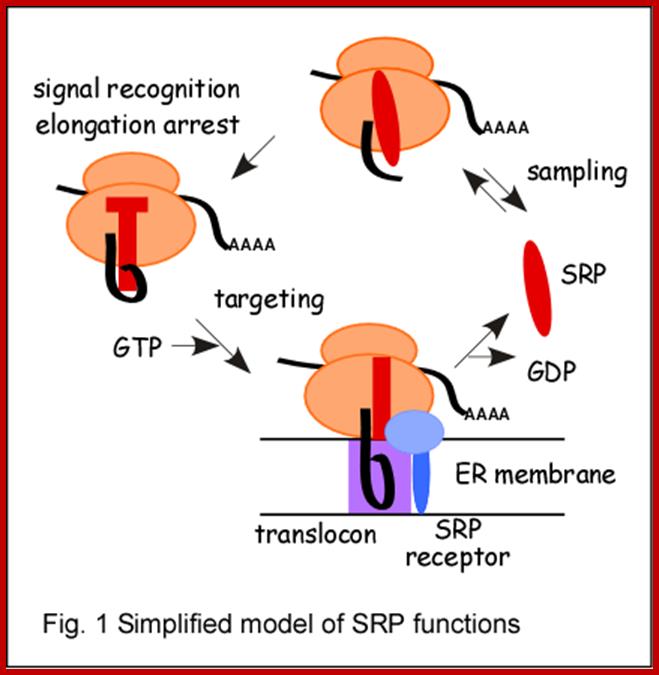
SRP mediated
protein targeting: The mammalian signal recognition particle (SRP) plays an
essential role in the biogenesis of proteins that have to insert or cross the
endoplasmic reticulum membrane (ER) membrane. It targets nascent chains to the
translocation apparatus in the ER and therefore provides a link between protein
synthesis and the nascent chain transfer into or across the ER membrane.
Translocation of proteins into the endoplasmic reticulum (ER) is the first step
in the secretory pathway of many luminal and membrane proteins as well as of
secretory proteins and is therefore vital in generating and maintaining
cellular structures and functions.
SRP acts in intimate relationship with another
ribonucleoprotein complex, the ribosome. The tight coupling between polypeptide
synthesis and translocation presumably precludes premature folding of
polypeptides in the cytosol. ;Katherine
Strub; http://cms2.unige.ch/
Tm RNA: In bacteria, often the ribosomal machinery, while translating comes across with an mRNA does not have TER sequences for transcriptional termination.
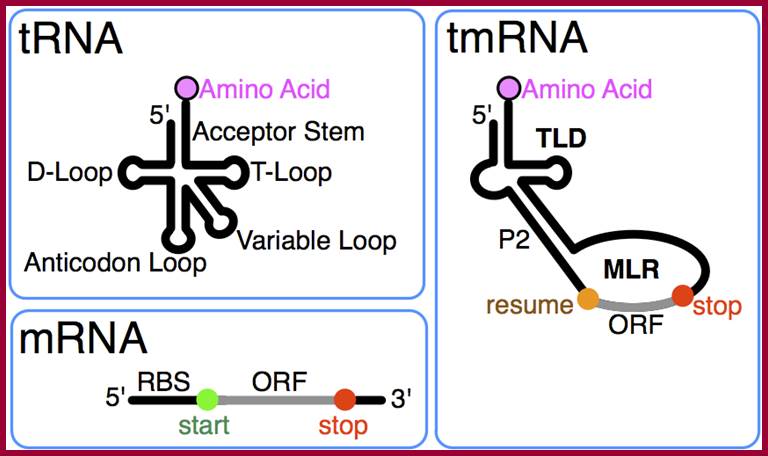
www.wikiwand.com
In such events ribosomes initiated translation gets stuck. A large number of ribosomes sequestered in such mRNAs. To avoid such situation prokaryotic systems have devised a mechanism where they are called tmRNA, which solves and salvages the problem. This RNA is 100 nucleotides long or more, but shows tRNA like structure at its 3’ end and the tRNA like structure is like Alanine tRNA. Towards its 5’end it contains at least ten Alanine coding sequences. The 3’ end of tmRNA gets loaded with Alanine and the same is placed at A site and a peptide bond is formed between the last amino acid stalled that was translating. At the same tine 5’ end of the tmRNA gets loaded on the stalled ribosomes in such a way the 5’ end sequences are read and translated and terminated with a ter-codon. The released protein, for having a number of Alanine are recognized and ubquitinated by ubiquitin complexes. Then they are fed to proteasome for degradation, thus such incorrect proteins are degrade for the released amino acids can be used for protein synthesis.
RNase P-RNA:
p RNA, is a Ribozymal RNA, capable performing endo-nucleolytically cutting and joining, but its efficiency increase with the binding of its associated protein subunits. Group I intron-RNAs is also capable of cutting and splicing Intronic RNA. Similar type RNAs are also found in eukaryotic systems, such as MRP RNAs (eukaryotic).
Structure of phi29 DNA packaging motor and packaging RNA.;
From Fabrication of pRNA nanoparticles to deliver therapeutic RNAs and bioactive compounds into tumor cells
(a) Bacteriophage phi29 DNA packaging motor. Six copies of pRNAs assemble into a hexamer ring to gear the viral DNA packaging motor. (b) The primary sequence and secondary structure of wild-type pRNA. The two domains (helical foot and central right- and left-hand loops) are connected by a 3WJ core. Reproduced with permission ; Yi Shu,Dan Shu, Farzin Haque; & Peixuan Guo; http://www.nature.com/
These RNAs are used in cleaving 5’ end of tRNAs to generate 5’ end of the tRNAs, a distinct feature of all tRNAs. These are 370 390 ntds long and have well define tertiary structure and they perform such process to generate 5’ end of the tRNAs.
Pi RNA and 21U RNA:
More recently, the discovery of two new species, viz., piRNA and 21-U RNA, has led to the addition to the existing list of small RNA classes. These were assigned to different small RNAs from the previously established classes on the basis of their origin and biogenesis. Nonetheless, they share some overlapping features shown by the previous small RNA classes as well.
Pi RNA:
piRNA biogenesis pathway. (a) Usually a polycistronic transcript, driven by mono- or bidirectional promoter, generates piRNAs by an unknown mechanism. Since the precursor lacks any tendency to achieve double-stranded form, the piRNA biogenesis seemed to be different from other small RNAs. (b) The biogenesis requires template to catalyze generation of desired small RNAs which further cleave corresponding target messages with another set of proteins. These piRNA may either regulate genome organization by checking transposon mobility or move to cytoplasm to take care of cognate messages either by cleaving or stabilizing them. Based on the mapping analysis of piRNA sequences onto genome it is postulated that these piRNA precursors are derived either from the non-overlapping transcripts generated from divergent promoter [175] or from a promoter giving rise to long single stranded RNA.

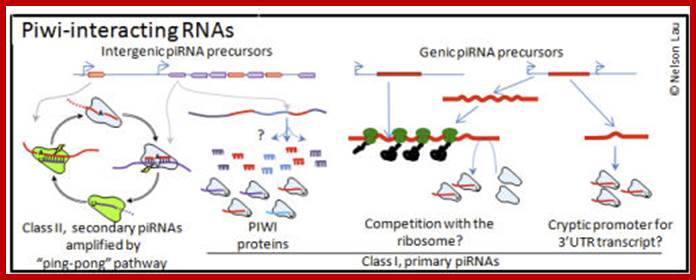
Small regulatory RNAs (and their associated proteins) are dynamic and active participants in controlling the genome and phenotype of cells and animals. Germ cells have a special role in the perpetuation of organisms, but they have also become the richest environment for the study of small regulatory RNAs. In addition to microRNAs, which are vital gene regulators conserved from plants to people, germ cells express endogenous small interfering RNAs (endo-siRNAs) and Piwi-interacting RNAs (piRNAs). http://www.bio.brandeis.edu/
Functional attributes:
Previous studies have shown that PIWI performs multiple functions ranging from epigenetic programming and repression of transposition to post transcriptional regulation. However, in contrast to negative PTGS regulation of si- and miRNAs, piRNAs promote stability of target mRNA and probably enhance the translation as well. Having loci spread throughout the genome, the most important role that could be conferred upon piRNAs would be the patronage of their respective loci. However, in view of the ability of piRC to cleave the cognate transcript, the involvement of piRNA at post-transcriptional level cannot be overruled
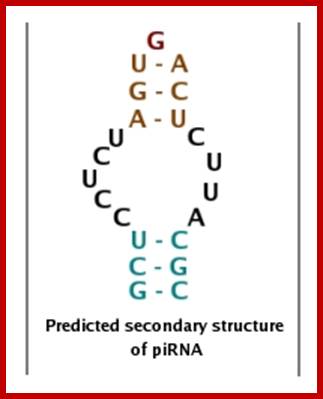
Predicted secondary structure of piRNA; piRNABank provides information- Simple search;Search piRNA clusters;Search homologous piRNAs;piRNA visualization map; Analysis tools;Deposited Sequences; http://pirnabank.ibab.ac.in/
piRNAs can be transmitted maternally, and based on research in D.melanogaster, piRNAs may be involved in maternally derived epigenetic effects. The activity of specific piRNAs in the epigenetic process also requires interactions between Piwi proteins and HP1a, as well as other factors

The biogenesis of piRNAs is not yet fully understood, although possible mechanisms have been proposed. piRNAs show a significant strand bias, that is, they are derived from one strand of DNA only[1], and this may indicate that they are the product of long single stranded precursor molecules. A primary processing pathway is suggested to be the only pathway used to produce pachytene piRNAs; in this mechanism, piRNA precurors are transcribed resulting in piRNAs with a tendency to target 5’ uridines. Also proposed is a ‘Ping Pong’ mechanism wherein primary piRNAs recognise their complementary targets and cause the recruitment of Piwi proteins. This results in the cleavage of the transcript at a point ten nucleotides from the 5’ end of the primary piRNA, producing the secondary piRNA. These secondary piRNAs are targeted toward sequences that possess an adenine at the tenth position[17]. Since the piRNA involved in the ping pong cycle directs its attacks on transposon transcripts, the ping pong cycle acts only at the level of transcription. One or both of these mechanisms may be acting in different species; C.elegans, for instance, does have piRNAs, but does not appear to use the ping pong mechanism at all.

Proposed pi RNA structure
piRNAs have been identified in both vertebrates and invertebrates, and although biogenesis and modes of action do vary somewhat between species, a number of features are conserved. piRNAs have no clear secondary structure motifs [1],.[4] the length of a piRNA is, by definition, between 26 and 31 nucleotides, and the presence of a 5’ uridine is common to piRNAs in both vertebrates and invertebrates. piRNAs in C.elegans have a 5’ monophosphate and a 3’ modification that acts to block either the 2’ or 3’ oxygen [5], and this has also been confirmed to exist in D.melanogaster[6], zebrafish [7], mice [8] and rats [7]. This 3’ modification is likely to be a 2’-O-methylation, but the reason for this modification is not known [7][9]. It is thought that there are many hundreds of thousands of different piRNA species found in mammals[10]. Thus far, over 50,000 unique piRNA sequences have been discovered in mice and more than 13,000 in D.melanogaster[11].
Location
piRNAs are found in clusters throughout the genome; these clusters may contain as few as ten or up to many thousands of piRNAs and can vary in size from one to one hundred kb. While the clustering of piRNAs is highly conserved across species, the sequences are not[1][13]. While D.melanogaster and vertebrate piRNAs have been located in areas lacking any protein coding genes, piRNAs in C.elegans have been identified amidst protein coding genes.
In mammals, piRNAs are found only within the testes, with an estimated one million copies per cell in spermatocytes and spermatids. In invertebrates, piRNAs have been detected in both the male and female germ lines, but in no other cell types.
At the cellular level, piRNAs have been found within both nuclei and cytoplasm, suggesting that piRNA pathways may function in both of these areas[3] and, therefore, may have multiple effects.

http://pirnabank.ibab.ac.in/
Proposed biogenesis of piRNA: miRNAs and siRNAs bind to AGO1 and AGO2 of the AGO subfamily proteins respectively. It has been identified in Drosophila that Aub and Piwi proteins bind with repeat-associated siRNA (rasiRNAs), which are produced from the antisense strand of retrotransposons in the germline. Sense rasiRNA precursors are produced by Slicer-like cleavage of the transcripts, which have a uracil (U) residue in their 5' end. The rasiRNAs do not require Dicer-1 or Dicer-2 for their biogenesis, which are essential for the production of siRNAs and miRNAs. Gunawardane et al, 2007, have proposed a model, wherein the primary antisense transcripts of rasiRNAs are cleaved by Slicing of the sense rasiRNAs associated with AGO3 proteins. Unlike Aub- and Piwi-associated rasiRNAs, those associated with AGO3 are produced from the sense strand of the retrotransposons. However, the AGO3-associated rasiRNAs complement their Aub and Piwi counterparts in the first 10 nucleotides and have a conserved adenine (A) residue at the 10th position. It has been suggested that rasiRNAs found in Drosophila are same as piRNAs identified in the mammalian germline (Haifan Lin., 2007). Figure 3 is a schematic of the proposed piRNA biogenesis model adapted from Gunawardane et al, 2007 and Haifan Lin, 2007
In the proposed piRNA biogenesis cycle, long single stranded piRNA precursors are processed by endonucleases, the nature of which is not yet elucidated. Slicer-like activity is observed in the Piwi subfamily proteins and hence, the Aub, Piwi and AGO3 proteins could possibly behave as endonucleases, involved in the piRNA biogenesis pathway. (Haifan Lin, 2007). http://pirnabank.ibab.ac.in/

miR-SNaREs , PIWI-SNaREs
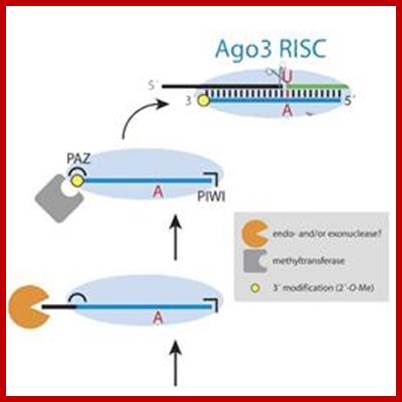
In addition to miRNAs and siRNAs, a third small RNA silencing system has been uncovered that prevents the spreading of selfish genetic elements. Production of the Piwi-associated RNAs (piRNAs), which mediate the silencing activity in this pathway, is initiated at a few master control regions within the genome. The nature of the primary piRNA-generating transcript is still unknown, but RNA interference (RNAi)-like cleavage events are likely defining the 5'-ends of mature piRNAs. We summarize the recent literature on piRNA biogenesis and function with an emphasis on work in Drosophila, where genetics and biochemistry have met very successfully; Julia Verena Hartig, Yukihide Tomari, and Klaus Förstemann ;http://www.cipsm.de

During gene transcription, RNA polymerase (Pol) II encounters obstacles, including lesions in the DNA template. Here, we review a recent structure–function analysis of Pol II transcribing DNA with a bulky photo-lesion in the template strand. The study provided the molecular basis for recognition of a damaged DNA by Pol II, which is the first step in transcription- coupled DNA repair (TCR). The results have general implications for damage recognition and the TCR mechanism. Ekaterina Kashkina, Michael Anikin, Florian Brueckner, Elisabeth Lehmann, Sergey N. Kochetkov, William T. McAllister, Patrick Cramer, and Dmitry Temiakov; J.Biol.chem 2007
Recent studies have reported the identification of piwi-associated RNAs (piRNAs) in Drosophila somatic cells. Interestingly, these piRNAs derive from the 30 untranslated regions of a subset of transcribed protein-coding genes and, experimentation suggests, might control the expression of other protein-coding transcripts. Studies of additional organisms support the new pathway’s presence across animals
In addition to miRNAs and siRNAs, a third small RNA silencing system has been uncovered that prevents the spreading of selfish genetic elements. Production of the Piwi-associated RNAs (piRNAs), which mediate the silencing activity in this pathway, is initiated at a few master control regions within the genome. The nature of the primary piRNA-generating transcript is still unknown, but RNA interference (RNAi)-like cleavage events are likely defining the 5'-ends of mature piRNAs. We summarize the recent literature on piRNA biogenesis and function with an emphasis on work in Drosophila, where genetics and biochemistry have met very successfully.
Piwi-interacting RNA (piRNA): It is the largest class of small RNA molecules that is expressed in animal cells. piRNA forms RNA-protein complexes through interactions with Piwi proteins. These piRNA complexes have been linked to transcriptional gene silencing of Retrotransposons and other genetic elements in germ line cells, particularly those in spermatogenesis. They are distinct from miRNA in size (26–31 nt rather than 21–24 nt), lack of primary sequence conservation, and increased complexity.
The wide variation in piRNA sequences and PIWI function over species contributes to the difficulty in establishing the functionality of piRNAs. However, like other small RNAs, piRNAs are thought to be involved in gene silencing, specifically the silencing of transposons. The majority of piRNAs are antisense to transposon sequences, suggesting that transposons are the piRNA target. In mammals it appears that the activity of piRNAs in transposon silencing is most important during the development of the embryo, and in both C.elegans and humans, piRNAs are necessary for spermatogenesis.
RNA Silencing:
piRNA has a role in RNA silencing via the formation of an RNA-induced silencing complex (RISC). piRNAs interact with Piwi proteins that are part of a family of proteins called the Argonautes. These are active in the testes of mammals and are required for germ-cell and stem-cell development in invertebrates. Three Piwi subfamily proteins - MIWI, MIWI2 and MILI - have been found to be essential for spermatogenesis in mice. piRNAs direct the Piwi proteins to their transposon targets. A decrease or absence of PIWI protein expression is correlated with an increased expression of transposons. Transposons have a high potential to cause deleterious effect on their hos, and, in fact, mutations in piRNA pathways are found to reduce fertility in D.melanogaster. However, piRNA pathway mutations in mice do not demonstrate reduced fertility; this may indicate redundancies to the piRNA system. Further, it is thought that piRNA and endogenous small interfering RNA (endo-siRNA) may have comparable and even redundant functionality in transposon control in mammalian oocytes.
piRNAs appear to have an impact on particular methyltransferases that perform the methylations which are required to recognize and silence transposons, but this relationship is not well understood.
Epigenetic Effects:
piRNAs can be transmitted maternally, and based on research in D.melanogaster, piRNAs may be involved in maternally derived epigenetic effects. The activity of specific piRNAs in the epigenetic process also requires interactions between Piwi proteins and HP1a, as well as other factors.
Recent discovery also show, the existence of snoRNA, microRNA, piRNA characteristics in a novel non-coding RNA: x-ncRNA and its biological implication in Homo sapiens.
21U RNA:
In an attempt to redefine the small RNA profile in C. elegans, Ruby et al. [22] encountered a novel class of small RNAs, viz., 21U-RNAs. In all the reads analyzed, these molecules were found to be exactly 21 nucleotides long with Uridine at its 5′ end. Of the ~5454 sequences obtained, majority were mapped to two major regions on chromosome IV, with few reads lying in between the major regions.
It was elucidated that these species are sensitive towards alkaline hydrolysis and phosphatase treatment and their capacity to act as substrate for RNA ligase confirms these to be RNA molecules. Similar to small RNAs in plants and rasiRNAs in flies, 21U-RNAs also seemed to be modified at either 2´ or 3´ oxygen [22]. Extrapolating such resemblance to the functionality of these entities suggest that they might play some role(s) in chromatin reorganization and genome stability. In the absence of any evidence for the existence of dsRNA precursor, the biogenesis of 21-U RNAs seems to be dependent on some factor(s), which could sense the Uridine residue as the reference point to count the bases. 21U-RNAs show no particular strand biasedness and majority were mapped to intergenic or intronic regions. Because authors used mixed-stage libraries, they could not conclude, as to which stage these species starts accumulating most, which would provide some clues of their functionality. The presence of 21U-RNAs during L1 and dauer stages suggests their role during worm development.
Taking closer look at the sequences flanking 21U-RNAs Ruby et al. predicted two upstream elements, large and small motifs. While the large motif are ~34 nt long with 8-nucleotide core consensus sequence CTGTTTCA, the small motifs were ~4 ntd long having YRNT as the core sequence. These two motifs were separated by linker sequences of ~19-27 bps in all the cases (Figure 8). Further analysis revealed that these motifs were highly conserved, suggesting for the requirement of these sequences during the transcription. In contrast, the 21U-RNA sequences were not at all conserved even within the same species.
Each 21U-RNA is transcribed autonomously suggests that they are independent genes and that 5′ flanking sequences may act as promoter clearly supports the hypothesis (Figure 9). The consideration of the above fact would dramatically influence the current scenario where it is believed that there are approximately 25,000 genes, at least in worms (these small RNAs are not yet reported in other organisms). This number may increase up to 1.5 times the existing figure.
General structure of 21U-RNA locus
![]()
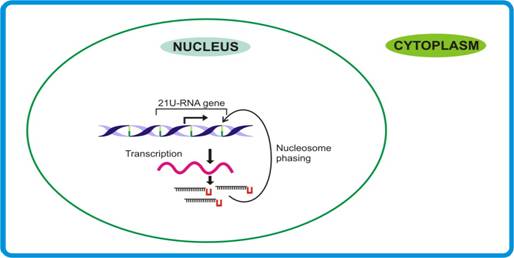
21U-RNA pathway. Dictated by their own promoters, the independent transcripts are made that may involve specific factors to sense the terminal U residue and the 21 nt, thereby releasing mature 21-U RNAs.
Function of 21-U RNAs
Considering the fact that 21-U RNA sequences show no homology with any transcript point towards their possible role in genome stability. However, in view of the earlier findings that sRNAs responsible for genome stability generally are of ≥24 nt, the possible role of 21U-RNAs (size 21nt) in genome stability remains doubtful. Moreover, since 21U-RNAs seemed to undergo maturation in the nucleus itself, their likely involvement in splicing cannot be over-ruled
Efference RNA:
Efference RNAs (eRNAs) are generally derived from eukaryotic intron sequences or they may derived from non-coding DNA sequences. Their functions is to regulate translational activity by interference with transcriptional apparatus or target proteins of the peptide in question. They can also provide a concentration based measure of protein expression. This is achieved by introducing fine-tined analog elements in gene regulation as opposed to the digital on-or-off regulation by promoters. This concept is very intriguing and fascinating and the knowledge of it is still meager.
Xist RNA: This is transcribed on one of the X chromosomal locus called Xic; the transcript substantially long, it cannot be translated and binds to X-chromosomal DNA , recruit methylases and prevents transcription thus makes all the genes in one of the two X chromosomes remain silent. The initial differential treatment of the two X chromosomes during X-chromosome inactivation is controlled by the X-inactivation center (Xic). This locus determines how many X chromosomes are present in a cell ('counting') and which X chromosome will be inactivated in female cells ('choice'). Critical control sequences in the Xic include the non-coding RNAs Xist and Tsix, and long-range chromatin elements. However, little is known about the process that ensures that X inactivation is triggered appropriately when more than one Xic is present in a cell. Using three-dimensional fluorescence in situ hybridization (FISH) analysis, we showed that the two Xics transiently colocalize, just before X inactivation, in differentiating female embryonic stem cells. Using Xic transgenes capable of imprinted but not random X inactivation, and Xic deletions that disrupt random X inactivation, we demonstrated that Xic colocalization is linked to Xic function in random X inactivation. Both long-range sequences and the Tsix element, which generates the antisense transcript to Xist, are required for the transient interaction of Xics. We propose that transient colocalization of Xics may be necessary for a cell to determine Xic number and to ensure the correct initiation of X inactivation.
At the
onset of mammalian X-chromosome inactivation, the X chromosomes are counted and
then a choice is made about which one to inactivate. New findings provide
evidence that a transient physical association between X chromosomes in the
nucleus might be involved in this process.
Figure. A map of known genes and regulatory
elements in the Xic region surrounding the Xist gene:

Figure. A map of known
genes and regulatory elements in the Xic region surrounding the Xist
gene. The Tsix transcript (brown arrow) is antisense
to Xist (green arrow), mainly initiates 16 kb
downstream of Xist and extends over at least 40 kb. The
DNA regulatory element Xite (purple) and the DXPas34 minisatellite (purple) have been shown
to enhance (+) Tsix expression16, which in turn
represses Xist (-). The structures of the 65-kb
deleted (65![]() ) and 16kb-complemented alleles, and the Xic YAC used by Bacheret al. are
shown below. The occurrence of X–X association in female-deleted cells and of
X-autosome association in male transgenic cells is indicated to the left. Celine Morey and Windy Bickmore http://www.nature.com
) and 16kb-complemented alleles, and the Xic YAC used by Bacheret al. are
shown below. The occurrence of X–X association in female-deleted cells and of
X-autosome association in male transgenic cells is indicated to the left. Celine Morey and Windy Bickmore http://www.nature.com
The Tsix transcript (brown arrow) is antisense to Xist (green arrow), mainly initiates 16 kb downstream of Xist and extends over at least 40 kb. The DNA regulatory element Xite (purple) and the DXPas34 minisatellite (purple) have been shown to enhance (+) Tsix expression16, which in turn represses Xist (-). The structures of the 65-kb deleted (65D) and 16kb-complemented alleles, and the Xic YAC used by Bacher et al. are shown below. The occurrence of X–X association in female-deleted cells and of X-autosome association in male transgenic cells is indicated to the left.
A time course of the events that initiate XCI during ES cell differentiation:
 A Timecourse of the events that initiate XCI
during ES cell differentiation. In undifferentiated ES
cells, transcription of Tsix (brown lines) from both chromosomes
maintains Xist expression at low levels and restrictsXist RNA to the transcription site (day 0).
Induction of differentiation triggers X–X colocalization, counting and choice. Tsix is then downregulated on the
presumptive Xi, thereby allowing Xist RNAs (blue lines) to accumulate in cis (day 2). On Xa, the
repression of Xist is maintained through the persistence
of Tsix expression promoted by the Xite locus. Subsequently, the Xi accumulates heterochromatic epigenetic
marks to maintain the silent state (day 4). Céline
Morey & Wendy Bickmore;http://www.nature.com/
A Timecourse of the events that initiate XCI
during ES cell differentiation. In undifferentiated ES
cells, transcription of Tsix (brown lines) from both chromosomes
maintains Xist expression at low levels and restrictsXist RNA to the transcription site (day 0).
Induction of differentiation triggers X–X colocalization, counting and choice. Tsix is then downregulated on the
presumptive Xi, thereby allowing Xist RNAs (blue lines) to accumulate in cis (day 2). On Xa, the
repression of Xist is maintained through the persistence
of Tsix expression promoted by the Xite locus. Subsequently, the Xi accumulates heterochromatic epigenetic
marks to maintain the silent state (day 4). Céline
Morey & Wendy Bickmore;http://www.nature.com/
In undifferentiated ES cells, transcription of Tsix (brown lines) from both chromosomes maintains Xist expression at low levels and restricts Xist RNA to the transcription site (day 0). Induction of differentiation triggers X–X co localization, counting and choice. Tsix is then down regulated on the presumptive Xi, thereby allowing Xist RNAs (blue lines) to accumulate in cis (day 2). On Xa, the repression of Xist is maintained through the persistence of Tsix expression promoted by the Xite locus. Subsequently, the Xi accumulates heterochromatic epigenetic marks to maintain the silent state (day 4).
X-inactivation seems to occur from an inactivation center
- Called XIC, mapped to Xq13
- The evidence is that if the X chromosome is broken by a translocation, only one of the two resulting segments undergoes inactivation. The location of the inactivation center has been mapped
XIST gene, encoded at site of X inactivation center:

http://faculty.virginia.edu/
XIC
- signal from X inactivation center, spread along chromosome, promoted by booster elements.
- signal spreading is less effective in autosomal material than in the X chromosome itself.
- LINEs may represent booster sequences
- X chromosome is strikingly LINE-rich.
- Efficiency of spread into autosomal regions correlates with their LINE content.
- inactive X replicates late, the late replication may help maintain the inactive signal, in which late replication prevents transcription and transcription is required for early replication.
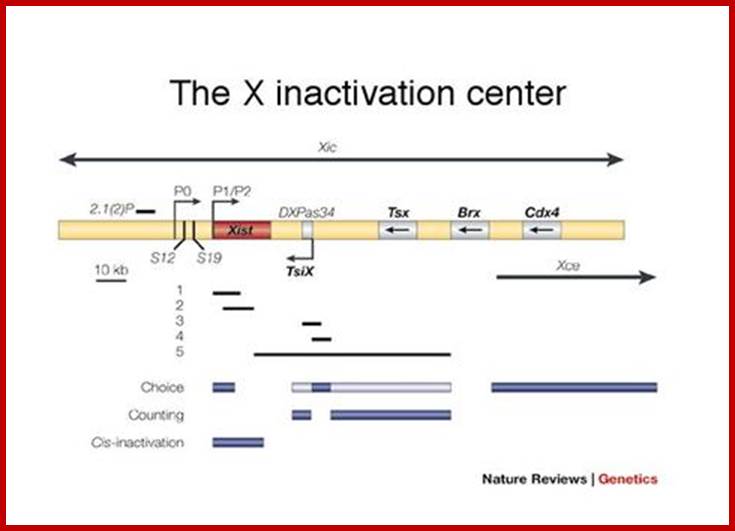
XIC;
- signal from X inactivation center, spread along chromosome, promoted by booster elements (Fig 3)
- signal spreading is less effective in autosomal material than in the X chromosome itself
- LINEs may represent booster sequences
- X chromosome is strikingly LINE-rich
- Efficiency of spread into autosomal regions correlates with their LINE content
- inactive X replicates late, the late replication may help maintain the inactive signal, in which late replication prevents transcription and transcription is required for early replication; ;http://faculty.virginia.edu/
Methylation may provide mechanism.
- signal may be methylation of the DNA, but methylation may be a consequence rather than a cause of the inactivation.
- differential methylation not found in marsupials, in which X inactivation always occurs on the paternal X .
Xist (mouse) and XIST (human):
- Xist= X Inactivation Specific Transcript
- pronounced e xist
- Xist is expressed only from the inactive X chromosome, not the active X chromosome
- Xist expressed after the 2 cell stage.
- Xist maps to the XIC
- has been cloned in mouse and human
Xist continued…
- RNA is expressed, but does not code for protein
- longest ORF (open reading frame).
- mouse 15 kb RNA, human 17 kb RNA
- RNA remains trapped in nucleus
- expressed only from the inactive X chromosome
- expressed in all mature female cells, and briefly in testes when an inactive X chromosome is present
- stabilization of Xist RNA mediates initiation of X inactivation
- XIST RNA may form a "cage" around the chromosome, acting as a barrier to RNA polymerases and blocking transcription
- The act of transcribing XIST RNA may induce a conformational change in the chromosome that inactivates it
- XIST RNA, alone or interacting with a nuclear factor, binds to the chromosome, inducing conformational change that inactivates it
- The act of transcribing XIST RNA "opens" a site on the X chromosome that allows it to attach to some inactivation machinery on the nuclear membrane
- to explain why Barr bodies are on the nuclear membrane
- XIST RNA, alone or interacting with a nuclear factor, binds to a site on the X chromosome that allows it to attach to some inactivation machinery on the nuclear membrane
Xist Mechanism:
- XIST RNA may form a "cage" around the chromosome, acting as a barrier to RNA polymerases and blocking transcription
- The act of transcribing XIST RNA may induce a conformational change in the chromosome that inactivates it
- XIST RNA, alone or interacting with a nuclear factor, binds to the chromosome, inducing conformational change that inactivates it
- The act of transcribing XIST RNA "opens" a site on the X chromosome that allows it to attach to some inactivation machinery on the nuclear membrane
- to explain why Barr bodies are on the nuclear membrane
- XIST RNA, alone or interacting with a nuclear factor, binds to a site on the X chromosome that allows it to attach to some inactivation machinery on the nuclear membrane; ;http://faculty.virginia.edu/
Tsix and Xist have a yin and yang relationship.
- Tsix is an antisense transcript to Xist, is transcribed through the Xist locus
- Tsix is expressed during the pre-inactivation period

DNA methylation is essential to maintain X chromosome inactivation; at CpG islands on the inactive X; DNA cytosine methylation inhibitors such as 5-azacytidine can reactivate X-linked genes; demethylation precedes transcription during the reactivation of HPRT by 5-azacytidine; in marsupials X-linked CpG islands are not heavily methylated, nor are these genes kept stably inactivated; DNA methyltransferase -/- transgenic mouse embryos are not able to maintain genomic imprinting, nor can they properly control Xist expression, which is required for the establishment of X inactivation; http://faculty.virginia.edu/
DNA methylation is essential to maintain X chromosome inactivation
- at CpG islands on the inactive X
- DNA cytosine methylation inhibitors such as 5-azacytidine can reactivate X-linked genes
- demethylation precedes transcription during the reactivation of HPRT by 5-azacytidine
- in marsupials X-linked CpG islands are not heavily methylated, nor are these genes kept stably inactivated
- DNA methyltransferase -/- transgenic mouse embryos are not able to maintain genomic imprinting, nor can they properly control Xist expression, which is required for the establishment of X inactivation

Methylation CpG islands can cause imprinted gene character, which can be transmitted to their offsprings.

www.mdpi.com
The phenomena of X inactivation and imprinting are associated
- In some cases imprinting is involved in the choice of X chromosome for inactivation, with the paternally derived X chromosome being preferentially inactivated.
- DNA methylation is known or believed to be involved in the mechanism in both cases.
- However, imprinting affects individual genes in different ways, although it appears global in the sense that it will affect the haploid complement of chromosomes derived from one parent. X chromosome inactivation involves coordinated regulation of a whole chromosome.
- For most genes there is no difference in expression whether the allele has been inherited from the mother or from the father
- but some genes are influenced by their parental origin. They have a 'imprint' of their gametic origin.
- Its occurrence in mammals has only recently been recognized, and was deduced from a number of different lines of research, including classical genetic studies, studies on X-inactivation, and the development of diploid parthenogenetic (gynogenetic, and androgenetic) embryos.
Imprinting is a phenomenon whereby the activity of some genes depends on the gender of origin.
- maternal allele but not paternal allele is expressed in some cases
- paternal but not maternal allele is expressed in other cases
- disease can occur if the normally expressed allele is mutant
- disease can occur if there is uniparental disomy
- an individual may receive two copies of a specific chromosome from one parent instead of one maternal and one paternal homolog
- if two copies of the chromosome with the non-expressed allele are inherited, that locus will be nonfunctional by this epigenetic mechanism
Mechanism of imprinting not clear:
- how are maternal and paternal alleles distinguished?
- must carry an imprint to signal the difference
- must be stably inherited for many rounds of DNA replication
- imprint must be erased when it passes through the germline
- a man may transmit a chromosome inherited from his mother
- a woman may transmit a chromosome inherited from her father
Genomic imprinting requires erasure of the imprint in the germ line:

The consequences of a translocation between an X chromosome and an autosome are variable. a | An autosomal tumour suppressor (gene A) can become inactivated if it is translocated to the inactive copy of the X chromosome. A gene that is normally located within transcriptionally active euchromatin on an autosome will become silenced if it moves by translocation to an area of the inactive X chromosome (Xi) in which it comes under the influence of the X-chromosome inactivation centre (XIC), which promotes the formation of transcriptionally silent heterochromatin and loss of gene expression. b | Conversely, an X-chromosome-linked gene (gene B) that is normally located on the Xi, and is therefore not expressed, can be reactivated by translocation to an autosome, with potential oncogenic consequences. In this situation, if the translocation separates the gene from the XIC, moving it to an area of transcriptionally active euchromatin on an autosome, the effects of heterochromatinization will be lost and the gene will be transcribed. Tumorigenic effects of translocations involving the X chromosome; Alain Spatz, Christophe Borg & Jean Feunteun http://www.nature.com/
Mechanisms of X-chromosome inactivation:

Figure . Transcription maps of the Xic/XIC regions in mouse and human (27). There are 11 genes in the mouse Xic region: Xpct, Xist, Tsx, Tsix, Chic1 (formerly, Brx), Cdx4, NapIl2 (formerly, Bpx), Cnbp2, Ftx, Jpx, and Ppnx. Protein coding genes are represented by yellow boxes. Four of the 11 genes, Xist, Tsix, Ftx, and Jpx, are untranslated RNA genes and represented by red boxes. Region B, a non-coding expressed domain, is represented by a striped box. All the genes identified in mouse are conserved in human, except Ppnx and Tsix. In human, however, Tsx has become a pseudogene. The human region is approximately three times larger than the mouse. Despite this major change in size, the order and orientation of genes is conserved in human and mouse, except for Xpct, which is at the same location but in the inverse orientation. A histone H3 lysine 9 dimethylation hotspot and H4 hyperacetylation are represented by blue and green boxes below the transcription map of the Xic region in mouse. Pillet et al. showed that the region -1157 to +917 has no in vitro sex-specific promoter activity. A minimal constitutional promoter was assigned to a region from -81 to +1. Deletion of the segment -441 to -231 is associated with an increase in CAT activity and may represent a silencer element. The choice/imprinting center contains tandem CTCF binding sites. Chao et al. proposed that Tsix and CTCF together establish a regulatable epigenetic switch for X-inactivation. Ogawa and Lee showed that Xite, located 10 kb from the Tsix transcription start, harbors two clusters of DNase hypersensitive sites. Samuel C. Chang; Samuel C. Chang, Tracy Tucker, Nancy P. Thorogood, and Carolyn J. Brown ; Department of Medical Genetics, University of British Columbia,Vancouver, BC, Canada
Figure: Accumulation of chromatin changes during X inactivation- The timing of changes to the inactive X are ordered as observed in studies of early mouse development and ES cell differentiation. Silencing can result from Xist expression, but stabilization of the silencing requires additional changes. Reactivation occurs rarely as discussed in the text, and in general the inactivation status is very stably maintained once established.Frontiers- http://www.bioscience.org

Figure: Chromatin remodeling on the inactive X chromosome. A. Active X chromatin is characterized by acetylation of H3 and H4 of the core nucleosome. There is also methylation of H3 lysine 4. B. Inactive X chromosome. Upon expression and localization of Xist there is macroH2A recruitment. It is unclear if these are bound together physically or are associated in some yet unidentified ribonuclear protein complex. The histone tails on the inactive X become hypoacetylated and methylated at H3 lysine 9 and 27, and H4 lysine 20. In addition, ubiquitination of H2A lysine 119 within the histone body is observed. DNA methylation is a late event in the inactivation process to lock in the inactive state. Note: it is not known to what extent the histone modifications are occurring on the same histone or within the same nucleosome, but at least some appear to be found in alternate domains . http://www.bioscience.org
Transposons:
Transposable elements (TEs), also known as "jumping genes," are DNA sequences that move from one location on the genome to another. These elements were first identified more than 50 years ago by geneticist Barbara McClintock of Cold Spring Harbor Laboratory in New York. Biologists were initially skeptical of McClintock's discovery. Over the next several decades, however, it became apparent that not only do TEs "jump," but they are also found in almost all organisms (both prokaryotes and eukaryotes) and typically in large numbers. For example, TEs make up approximately 50% of the human genome and up to 90% of the maize genome (SanMiguel, 1996).
Types of Transposons
Today, scientists know that there are many different types of TEs, as well as a number of ways to categorize them. One of the more common divisions is between those TEs that require reverse transcription (i.e., the transcription of RNA into DNA) in order to transpose and those that do not. The former elements are known as retrotransposons or class 1 TEs, whereas the latter are known as DNA transposons or class 2 TEs. The Ac/Ds system that McClintock discovered falls in the latter category. Different classes of transposable elements are found in the genomes of different eukaryotic organisms (Figure 1)
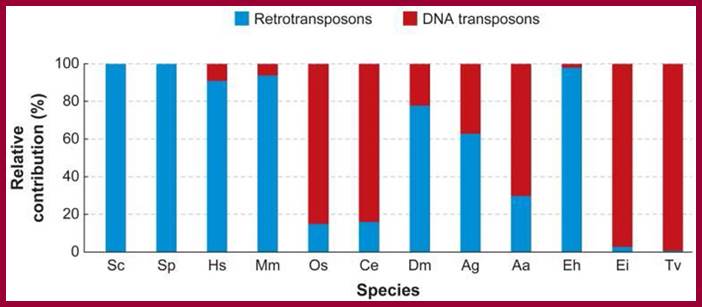
Figure ; The relative amount of retrotransposons and DNA transposons in diverse eukaryotic genomes
This graph shows the contribution of DNA transposons and retrotransposons in percentage relative to the total number of transposable elements in each species. (Sc: Saccharomyces cerevisiae; Sp: Schizosaccharomyces pombe; Hs: Homo sapiens; Mm: Mus musculus; Os: Oryza sativa; Ce: Caenorhabditis elegans; Dm: Drosophila melanogaster; Ag: Anopheles gambiae, malaria mosquito; Aa: Aedes aegypti, yellow fever mosquito; Eh: Entamoeba histolytica; Ei: Entamoeba invadens; Tv: Trichomonas vaginalis.)
© 2007 Annual Reviews Feschotte, C. & Pritham, E. J. DNA transposons and the evolution of eukaryotic genomes. Annual Reviews in Genetics 41, 331–348. All rights reserved
DNA Transposons;
All complete or "autonomous" class 2 TEs encode the protein transposase, which they require for insertion and excision (Figure 2). Some of these TEs also encode other proteins. Note that DNA transposons never use RNA intermediaries—they always move on their own, inserting or excising themselves from the genome by means of a so-called "cut and paste" mechanism.

Classes of mobile elements.
DNA transposons (e.g., Tc-1-mariner) have inverted terminal inverted repeats (ITRs) and a single open reading frame (ORF) that encodes a transposase. They are flanked by short direct repeats (DRs). Retrotransposons are divided into autonomous and nonautonomous classes depending on whether they have ORFs that encode proteins required for retrotransposition. Common autonomous retrotransposons are (i) LTRs or (ii) non-LTRs. Examples of LTR retrotransposons are human endogenous retroviruses (HERV) (shown) and various Ty elements of S. cerevisiae (not shown). These elements have terminal LTRs and slightly overlapping ORFs for their group-specific antigen (gag), protease (prt), polymerase (pol), and envelope (env) genes. They produce target site duplications (TSDs) upon insertion. Also shown are the reverse transcriptase (RT) and endonuclease (EN) domains. Other LTR retrotransposons that are responsible for most mobile-element insertions in mice are the intracisternal A-particles (IAPs), early transposons (Etns), and mammalian LTR-retrotransposons (MaLRs). These elements are not present in humans, and essentially all are defective, so the source of their RT in trans remains unknown. L1 is an example of a non-LTR retrotransposon. L1s consist of a 5'-untranslated region (5' UTR) containing an internal promoter, two ORFs, a 3' UTR, and a poly(A) signal followed by a poly(A) tail (An). L1s are usually flanked by 7- to 20-bp target site duplications (TSDs). The RT, EN, and a conserved cysteine-rich domain (C) are shown. An Alu element is an example of a nonautonomous retrotransposon. Alus contain two similar monomers, the left (L) and the right (R), and end in a poly(A) tail. Approximate full-length element sizes are given in parentheses.
© 2004 American Association for the Advancement of Science Kazasian, H. H. Mobile elements: drivers of genome evolution. Science 303, 1626–1632 (2004). https://www.nature.com
Leslie A. Pray, Ph.D. © 2008 Nature Education ; https://www.nature.com
Telomerase RNA:
Telomeres are the structures found at the ends of euchromatin chromosomes for they are linear. The telomeres can be observed under proper staining and high resolution microscopes as fine granular structures. The Telomeric DNA consists of some repeated sequences. Such sequences and such elements were firs observed in Tetrahymena thermophillis animal. Since then, it was found that all almost all eukaryotic chromosomes have such Telomeric sequences at the chromosomal ends without any exceptions. In humans the repeat sequences are GGGATT. This sequence can be repeated several hundred times. This length can shrink or expand. Active cells have longer Telomeric ends and aged cells have shortened Telomeric ends. The replication chromosomal ends have problem for the 5’ends for they don’t have primers to fill, thus the ends remain as free ends. Such ends are subjected 5’exonucleases. To protect such ends the 5’ end id elongated by special mode of replication and it gets folded into a compact structure with the association of telomere specific proteins. For elongation of 5’ end tails, an RNA called Telomeric RNA, and the same is used which base pairs with the ends and it is copied by eukaryotic Reverse transcriptase. Thus the ends retraction is prevented and extended.

pRNA:
Promoter RNA called pRNAs correspond to promoter regions and such RNAs can act as scaffold to bind to the antisense strand of the promoter and direct siRNAs resulting in epigenetic remodeling. This amounts siRNA directed transcriptional gene silencing in human calls.
dsRNA:
These ds RNAs are produced within a cell and they act as activators of several effector molecules such as specific protein kinases (PKR), RNase L activation through 2’, 5’oligos synthesis, activate few nuclear RNase III.

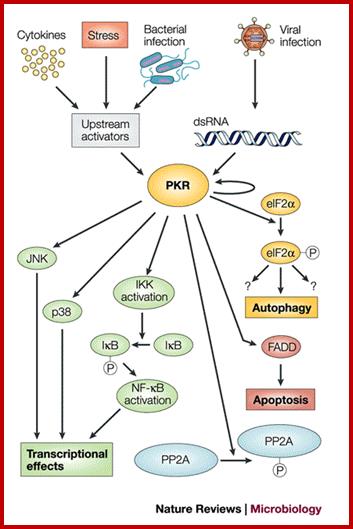
Immune responses to dsRNA: Implications for gene silencing technologies; dsRNA can mobilize several intracellular and extracellular antiviral mechanisms that have broad ranging cellular effects. Once activated, two distinct mechanisms, dsRNA-associated protein kinase (PKR) and oligoadenylate synthetase (OAS), function to shut down protein translation. In addition, gene silencing mechanisms, such as RNA interference (RNAi), leads to the degradation of complementary mRNA when dsRNA activates this pathway. and further, induction of IFN by dsRNA leads to the upregulation of the IFN-stimulated genes (ISG). These genes further promote translational inhibition by increasing proteins such as PKR and OAS, as well as generating an inflammatory response in the organism. The extracellular receptor toll-like receptor (TLR)3 also binds dsRNA and promotes the ISG., Tim J Doran and Andrew GD Bean; http://www.nature.com/; Response to dsRNA; Tim J Doran et asl; http://www.nature.com/
Ds RNA activated PKR has extensive action such as eIF2, inducing apoptosis, transcriptional effects through p-lation to JNK (kinase), NFkB. These ds RNA are synthesized or produced in response to bacterial, viral and even stress induction.
 Endogenous triggers of RNAi pathway include
foreign DNA or double-stranded RNA (dsRNA) of viral origin, aberrant transcripts from repetitive sequences in
the genome such as transposons, and
pre-microRNA (miRNA). In plants, RNAi forms the basis of virus-induced gene silencing (VIGS), suggesting an important role in pathogen resistance.
Endogenous triggers of RNAi pathway include
foreign DNA or double-stranded RNA (dsRNA) of viral origin, aberrant transcripts from repetitive sequences in
the genome such as transposons, and
pre-microRNA (miRNA). In plants, RNAi forms the basis of virus-induced gene silencing (VIGS), suggesting an important role in pathogen resistance.
A possible mechanism underlying the regulation
of endogenous genes by the RNAi machinery was suggested from studies of C.
elegans.
In mammalian cells long (>30nt)
double-stranded RNAs usually cause Interferon
response.
;Consequences of long dsRNAs in the nucleus and cytoplasm of mammalian cells; https://www.ncbi.nlm.nih.gov

A simplified model for the RNAi pathway is based on two steps, each involving RNAse enzyme. In the first step thetrigget RNA (either ds RNA or mi RNA transcript is processed into an short, interfering RNA (siRNA) by the RNase II enzymes Dicer and Drosha. In the second step, siRNAs are loaded into the effector complex RNA-induced silencing complex (RISC). The siRNA is unwound during RISC assembly and the single-stranded RNA hybridizes with mRNA target. Gene silencing is a result of nucleolytic degradation of the targeted mRNA by the RNase H enzyme Argonaute (Slicer). If the siRNA/mRNA duplex contains mismatches the mRNA is not cleaved. Rather, gene silencing is a result of translational inhibition; https://www.ncbi.nlm.nih.gov
Antisense RNA within the cytoplasm can activate several pathways. Two major pathways implicate PKR and 2’,5’ oligoA synthetase activation. PKR is activated through binding to dsRNA. Activated PKR phosphorylates eIF2α and IκB which results in translation blockage and IFN secretion respectively. Activated 2’,5’ oligoA synthetase generates oligoadenylates which in turn activate RNAse L. RNAse L can degrade both cellular and viral ssRNAs and dsRNAs. General nuclear pathways involve ADAR activation and RNAse-III-like induction. Long dsRNAs (>100 bp) that activate the ADAR protein family will be deaminated on A residues and completely unwound. These RNAs will be retained in the nucleus and degraded. Short dsRNAs (30-100 bp) will only be partially deaminated, exported to the cytoplasm and can be translated into altered proteins since I’s are decoded as G’s. Molecular tools for potential therapeutics; http://theses.ulaval.ca/
Very often some of the dsRNAs either long or small RNA hairpin like one end up in the formation of si or miRNAs which bind to specific mRNA at their terminal regions and induce enzyme for degradation.
Type I IFN induction, signaling and action. Left panel: dsRNA, a characteristic by-product of virus replication, leads to activation of the transcription factors NF- n B, IRF-3 and AP-1 (not shown). The cooperative action of these factors is required for full activation of the IFN- h promoter. IRF-3 is phosphorylated by the kinases IKK ( and TBK-1 which in turn are activated by the RNA-sensing complex of RIG-I, MDA5 and IPS-1/MAVS. A second signaling pathway involves endosomal TLR-3 and TRIF. Right panel: Newly synthesized IFN- h binds to the type I IFN receptor (IFNAR) and activates the expression of numerous ISGs via the JAK/STAT pathway. IRF-7 amplifies the IFN response by inducing the expression of several IFN- h subtypes. SOCS and PIAS are negative regulators of the JAK- STAT pathway. Mx, ISG20, OAS and PKR are examples of proteins with antiviral activity.

Type I IFN induction, signaling and action. Left panel: dsRNA, a characteristic by-product of virus replication, leads to activation of the transcription factors NF- n B, IRF-3 and AP-1 (not shown). The cooperative action of these factors is required for full activation of the IFN- h promoter. IRF-3 is phosphorylated by the kinases IKK ( and TBK-1 which in turn are activated by the RNA-sensing complex of RIG-I, MDA5 and IPS-1/MAVS. A second signaling pathway involves endosomal TLR-3 and TRIF. Right panel: Newly synthesized IFN- h binds to the type I IFN receptor (IFNAR) and activates the expression of numerous ISGs via the JAK/STAT pathway. IRF-7 amplifies the IFN response by inducing the expression of several IFN- h subtypes. SOCS and PIAS are negative regulators of the JAK- STAT pathway. Mx, ISG20, OAS and PKR are examples of proteins with antiviral activity. https://www.researchgate.net
RNAs act like switches called Riboswitches:
Riboswitches are very unique kind of controlling elements, which operate either at transcriptional level or translational level. The molecules involved in such control are again RNA molecules. Such Riboswitch RNAs found in bacteria, Achaea, fungi and plants, perhaps they may operate even in higher system, for the mechanism is vivacious and dynamic, which responds to the cellular environment inside or outside the cell and to the needs of the cell.
Few examples will suffice to justify for the existence of Riboswitches. E.coli is a highly proliferative bug, the darling of molecular biologists, is used for much innovative experimentation that led to great many discoveries. The RNA polymerase, a complex of 5 protein subunits is highly productive when there is sufficient amount of nucleotide triphosphates. But when the concentration of these nucleotides falls below certain level the polymerase become very unstable. The instability does not involve any proteins. Similarly the concentration methionine controls the activity of genes responsible for methionine synthesis. The levels of uncharged tRNAs control expression of aminoacyl tRNA synthetase. Many of the gene expression is regulated by the availability of some metabolites including vitamins such as Vitamin B12, TPP 9thiamine pyrophosphate), FMN (Flavin mononucleotide), Lysine, Guanine and Adenines and many more such metabolites. Some operate at transcriptional level and some at translational level. But all operate at RNA structural level. So Riboswitches are regulatory RNAs. They act as sensors of internal milieu of metabolites and control either at transcription or translation.
In Bacillus subtilis, the gene involved in the synthesis of Methionine is controlled at transcription level. The 5’ region of 200-nucleotide long leader sequence generates alternate stem loop structures; one acts as transcriptional terminator another acts as transcriptional anti terminator. This depends upon the availability of methionine. Logic is simple; when the methionine is available there is no need for the synthesis of the enzyme by the way of transcription and translation. When the methionine is in plenty, the S-Adenosyl methionine binds to terminator stem loop of the mRNA at 5’ UTR sequence, thus the transcription is prematurely terminated. When it is required transcription is allowed to proceed.
Similarly the levels of charged or uncharged tRNAs control the synthesis of amino acyl tRNA synthetases. When charged tRNAs for a specific amino acid is available there is no need for the synthesis of that aminoacyl-tRNA synthetase, it is waste. But when one finds uncharged tRNAs for that amino acids are found, it means the cell requires an enzyme that can charge tRNAs with amino acids for the proteins to be synthesized. In this case a 200-300 nucleotide long 5’ terminal region of the transcript with its secondary structure interacts and recognizes it cognate uncharged tRNA. The secondary structure that develops is transcription terminator structure. The interaction of uncharged tRNA with this structure, virtually like codon and anticodon, recognition through base pairing, induces a structural change that it no more acts like a transcriptional terminator and allows transcription to progress so it generates a transcript that produces the enzyme required for charging the tRNA. Perhaps these Riboswitches have global operations.
These small RNAs with secondary structure provide binding sites for certain organic molecules, which on binding bring about regulation gene expression or regulation RNA synthesis or function.

C. David Allis; ;http://librarynews.rockefeller.edu/

http://core.kmi.open.ac.uk/
Thermosensor Boxes:

T-box RNAs (see figure, part b) are normally found in the 5' UTR of genes that encode AMINOACYL-tRNA SYNTHETASES or related amino-acid-biosynthesis genes of Gram-positive organisms. Each folds into a structure that selectively recognizes a specific tRNA76. Recognition is guided largely through the formation of Watson–Crick base pairing between the T-box RNA and the anticodon loop of the target tRNA. Gene expression is activated in the presence of tRNAs that are not aminoacylated, thereby boosting the expression of genes that are needed to maintain an adequate pool of charged tRNAs. The T-box associated with the tyrS gene of B. subtilis (shown in the figure) functions via a transcription-termination mechanism. The T-box RNA rejects its matched tRNA when the tRNA is already aminoacylated. This permits formation of the terminator stem and subsequently downregulates gene expression (not shown). But, if the tRNA is not aminoacylated (as depicted in the figure), it is bound by the T-box and this permits expression of the adjoining synthetase gene by formation of an anti-terminator structure.; Maumita Mandal & Ronald R. Breaker ; http://www.nature.com/
Gene regulation by Riboswitches
Maumita Mandal & Ronald R. Breaker:
Classical mechanisms of bacterial gene control that involve mRNA structures require the ribosome (transcription attenuation) or protein factors. Transcription attenuation of the E. coli trp operon makes use of the process of translation to determine the levels of tryptophan in the cell. A sufficient supply of tryptophan allows the cell to produce a level of tryptophan-charged tRNA such that the ribosome rapidly transcribes a tryptophan-rich leader peptide (trpL). The ribosome sequesters anti-terminator sequences and, therefore, transcription of the remaining mRNA chain is terminated (as shown in the figure, part a). If tryptophan-charged tRNAs are rare, then the ribosome transcribes the leader more slowly and permits formation of the anti-terminator stem (not shown). The result of this is transcription of the full-length mRNA and expression of the operon, which encodes genes that are needed to boost the tryptophan concentration.
More small RNAs;
stRNA: Small temporal RNAs: A good example of such RNAs are let-4 and let-7 in C.elegans.
Now transcription initiation RNA (tiRNA) have been found. Some ncRNAs are large, like lincRNAs (large intervening non-coding RNAs), but most are small between 18 and 31 nt. Within in the small ncRNA group are piwi-interacting (piRNA), repeat associated small interfering (rasiRNA), Trans-acting short interfering RNAs (tasiRNA),

Three known mechanisms of riboswitch action upon binding of metabolite (M): a) Transcription termination. b) Inhibition of translation initiation. c) Auto-cleavage; Colettedavis;http://www.scq.ubc.ca/