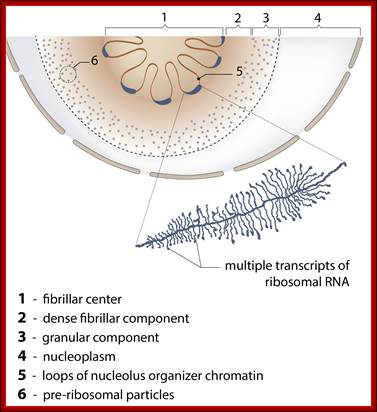
rRNA Processing in Eukaryotes:
Eukaryotic cells contain different types of RNAs. The list shows the kinds of them.

Classes of Eukaryotic RNAs:
- Ribosomal RNAs,
- Transfer RNAs,
- Messenger RNAs,
- Small molecular weight RNAs (80 or more kinds),
SnRNAs, Sc RNAs, Sno RNAs, mi/Si RNAs etc
A modular approach to cellular functions; http://www.mechanobio.info/
Chromatin loops share transcription factories: Chromatin loops from the same or from different chromosome territories often share transcription factories (TFs). TFs are enriched in RNA polymerase II complexes that produce nascent RNA transcripts, using co-transcribed genes from chromatin loops as their templates. The nucleolus may also anchor specific chromatin loci. In addition to rRNA genes, it often harbors large genomic regions (median size 750 kb) that are enriched in centromeric satellite repeats and inactive gene clusters. Centromeric regions are also found associated with nuclear lamina, suggesting that centromeres are distributed between the nuclear lamina and nucleoli. Molecular factories exist throughout every cell, persistently and efficiently synthesizing molecular components, proteins and lipids to keep up with the demands of cellular life. From the genetic blueprint contained within DNA, through to a perfectly folded and functional protein, cells must produce it all with the highest accuracy – lest defects in these products prove fatal for the cell, or its organism.
https://www.nobelprize.org
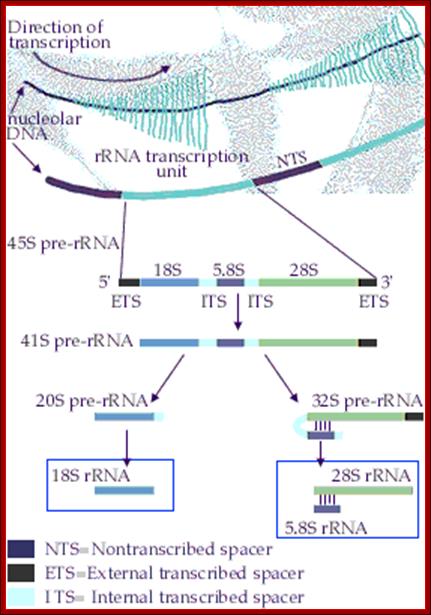
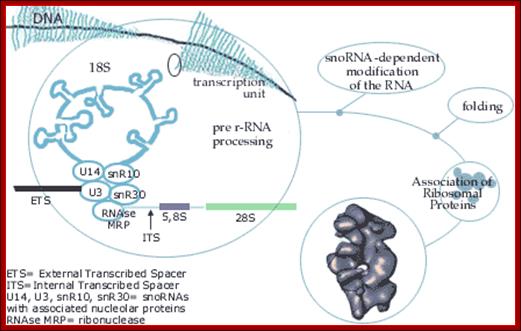
Top fig. rRNA synthesis and processing; The primary transcript is processed into the mature 18S, 5.8S and 28S rRNAs. The processing involves exo- and endo-nucleolytic cleavages guided by snoRNA (small nucleolar RNAs) in complex with proteins. The mature rRNAs contain modified nucleotides which are added after transcription by a snoRNA-dependent mechanism.
Bottom Fig. rRNA Transcripts appear as Christmas tree, the transcripts are cut and processed by snoRNAs; each transcript has ETS and ITS; U14,U3,snR10,snR30sboRNAswith associated nuclear proteins are involved in processing, PNase-ribonuclease. http://www.nobelprize.org/ educational;
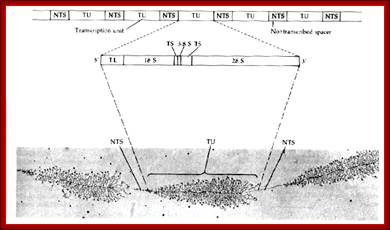
Organization of the genes for 18S, 5.8S, and 28S ribosomal RNA in amphibians. Each transcription unit (TU) is flanked by nontranscribed spacer regions (NTS). The transcription unit contains a transcribed leader sequence (TL), the 18S rRNA gene, a transcribed spacer (TS), the 5.8S rRNA gene, another transcribed spacer, and the gene for 28S rRNA. Beneath the diagram is an electron micrograph of tandemly arranged rRNA genes actively transcribing in the newt oocyte. The RNA gets progressively larger as it is transcribed, and dozens of transcripts are made simultaneously. (Photograph courtesy of O. L. Miller, Jr.); Scott F. Gilbert; http://9e.devbio.com/
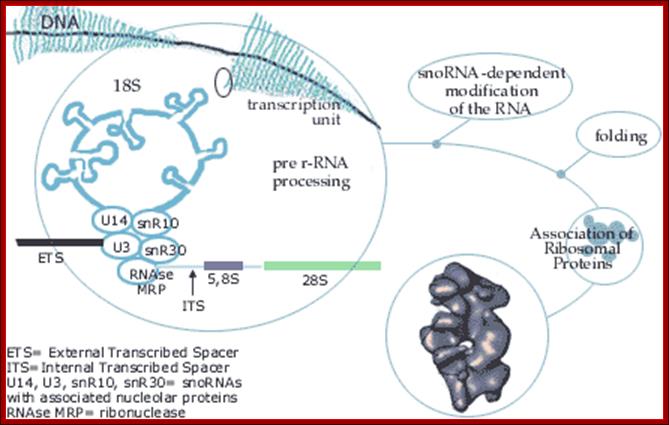
Processing involves two types, one modification of nucleotides and another ribose methylation; a set of Sno RNAs and their associated proteins are involved in the process. As they are modified they are cleaved endonucleolytically at specified positions determined by their secondary structure, and the fragments are further processed by 3’ and 5’ exonucleases.
Nucleolus (red) within Nucleus (red border); http://palaeos.com/http://www.biologyreference.com/
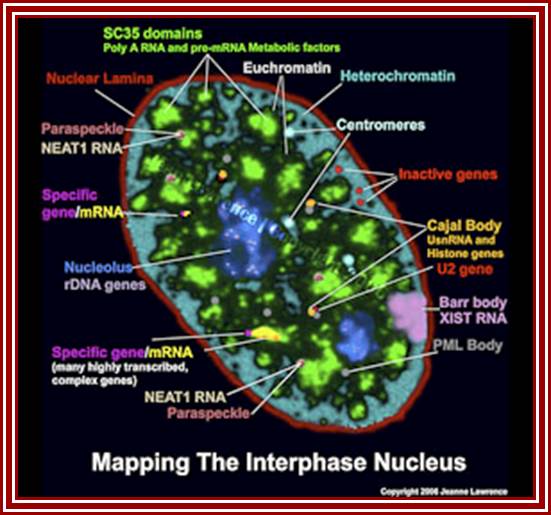
Nuclear and Genome Organization in Development –of a cell; Neat RNA is an architectural RNA that scaffolds a large nuclear structure. Neat RNA is a non-coding RNA that is required for the formation of paraspeckles (see image below). Paraspeckles are ubiquitous nuclear structures (~10-30/nucleus) of unknown function found in all human primary and transformed cells. http://www.umassmed.edu/
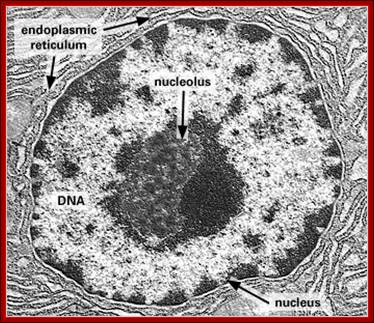
Dark stained Nucleolus within the Nucleus; http://www.frontiers-in-genetics.org/
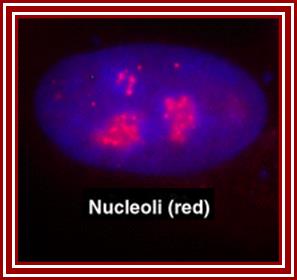
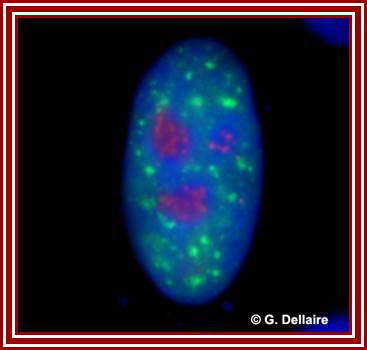
The red specks are the nucleoli found in the nucleus-fluorescent labeled Nucleolus; G.Dllaire; http://dellairelab.medicine.dal.ca
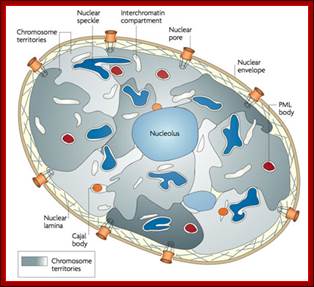 http://cellbiology.med.unsw.edu.au/units/images/nucleus_cartoon3.jpg
http://cellbiology.med.unsw.edu.au/units/images/nucleus_cartoon3.jpg
{kind=link}
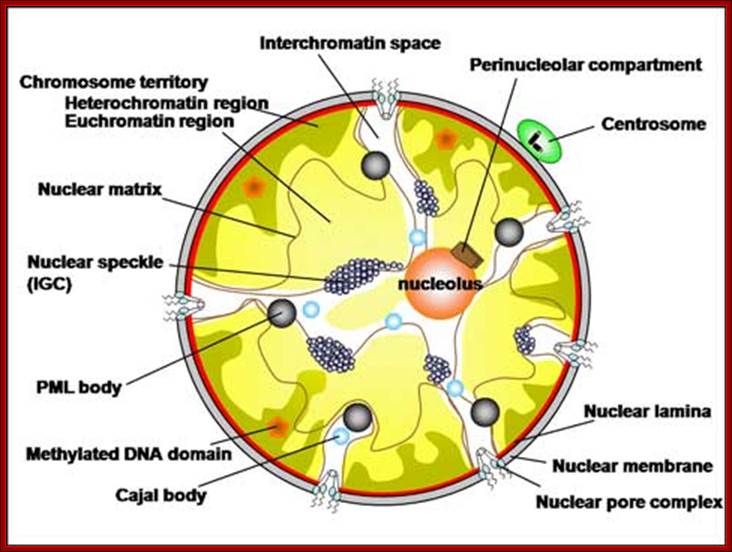
Hypothesized
models of nuclear structures.
Many specialized structures exist in the nucleus, and these structures are
assumed to provide the basis for various nuclear functions. Details of the
components, dynamics, and functions of the nuclear structures are uncertain;
Hitoshi Nishijima
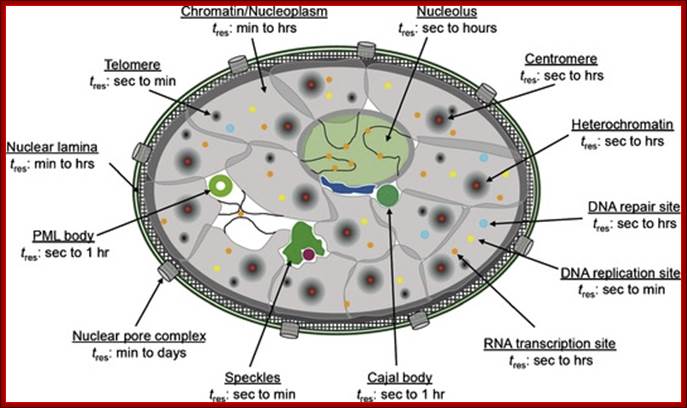
UNSW Cell Biology; (Im)mobilities in the nucleus. Illustration of subnuclear compartments and overview of their component‘s residence times (t res) determined by FRAP; Peter Hemmerich; http://cellbiology.med.unsw.edu.au/
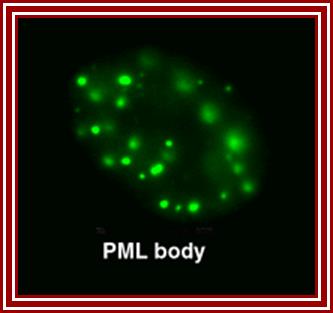
http://npd.hgu.mrc.ac.uk/
Five to thirty nuclear PML promyelotic leukemia, bodies are found, 0.2-1um accumulate specific proteins involved in apoptosis, senescence, gene regulation, tumour suppression etc.
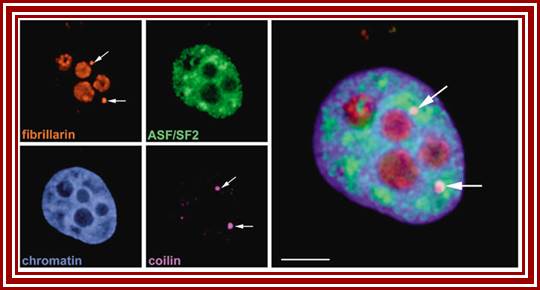
Left top-fibrillarin; next-ASF factors; bottom left-chromatin within the nucleus; next–coilin threads in the nucleus; right Nucleoli.
Nuclear speckles: Another spherical nuclear bodies little larger than Cajal bodies are also found in the nuclear sap and they contain nuclear RNA processing components such as p80/Coilins. They are enriched in pre-messenger RNA splicing factors and are located in the interchromatin regions of the nucleoplasm of mammalian cells. Speckles are dynamic structures, and both their protein and RNA-protein components can cycle continuously between speckles and other nuclear locations, including active transcription sites. They appear as irregular punctate structures of variable sizes
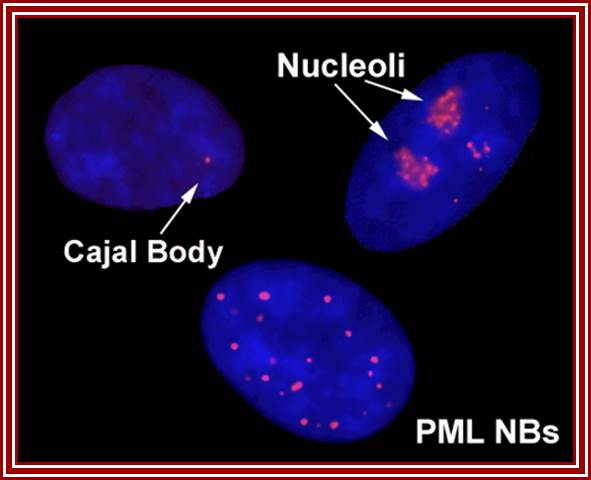
Cajal bodies, PML NBs = promyelocytic leukemia nuclear body (PML NB). https://blogs.dal.ca/
![[caption below]](Ribose_Nucleic_Acid2C-rRNA_Processing_in_Eukaryotes_files/image017.jpg)
http://jcb.rupress.org

Nuclear Speckles are dynamic structures and both their protein and RNA-protein components can cycle continuously between speckles and other nuclear locations, including active transcription sites. Speckles form in the interchromatin space. HeLa cells showing splicing factors localized in a speckled pattern as well as being diffusely distributed throughout the nucleoplasm. Bar = 5 µm. http://cshperspectives.cshlp.org
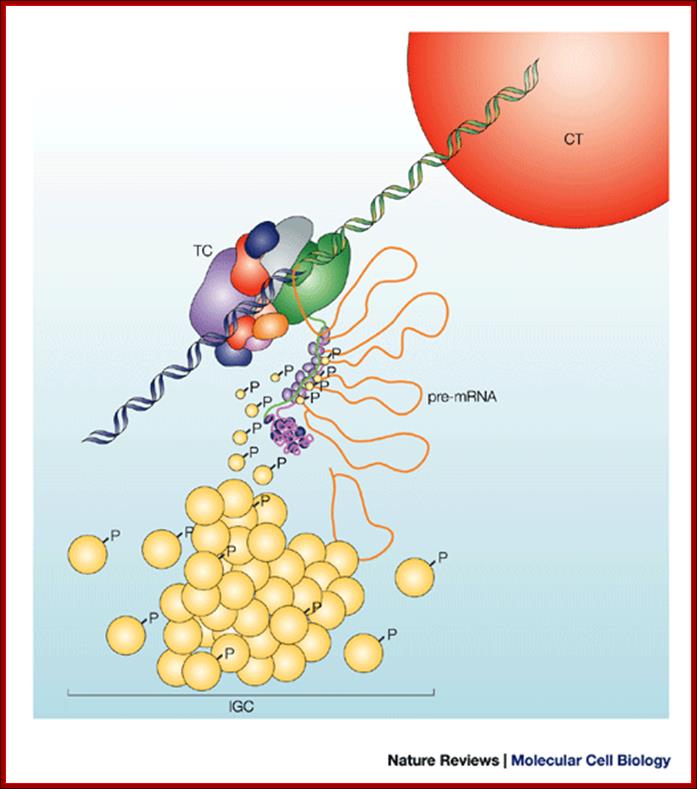
Nuclear speckles (interchromatin granule clusters; IGCs) form as the result of protein–protein interactions among pre-messenger RNA splicing factors and other constituents at the telophase/G1-phase transition. A basal level of factor exchange occurs between the speckles and the nucleoplasmic pool that is regulated by phosphorylation/dephosphorylation in a cell-type-specific manner. Modulation of the phosphorylation level of speckle proteins results in an increased release and recruitment to transcription sites. The model is not drawn to scale, and is modified with permission from Ref. 97 © Saunders (2002). CT, chromosome territory; IGC, interchromatin granule cluster; TC, transcription complex; pre-mRNA, pre-messenger RNA; Nuclear speckles: a model for nuclear organelles; Angus I. Lamond and David L. Spector.
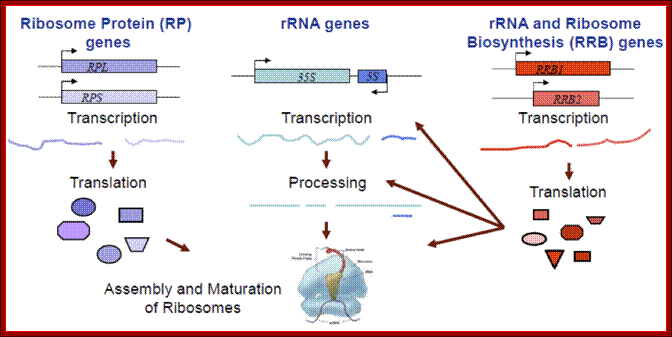
Ribosomal biogenesis requires the coordinated regulation of three extensive gene networks including 137 cytoplasmic genes and 150 rRNA genes and some 200 RRB genes; Michael Mcalear; mmcalear.faculty.wesleyan.edu
Ribosomal Subunits and their components:
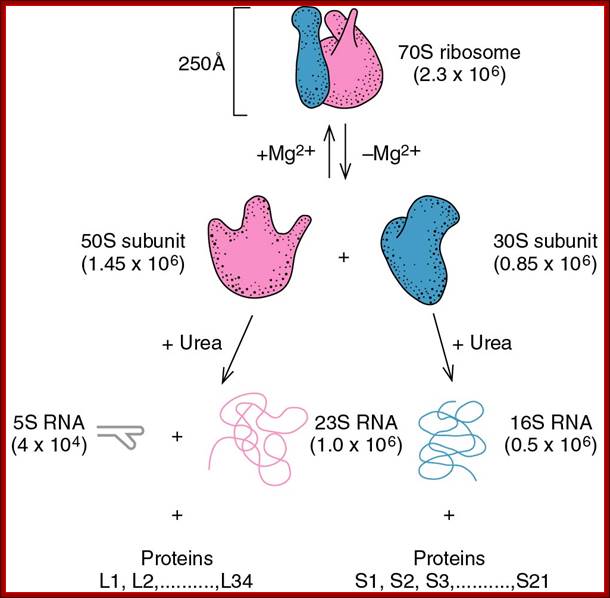
70s ribosomal subunits and their rRNA and proteins; http://biology.kenyon.edu/
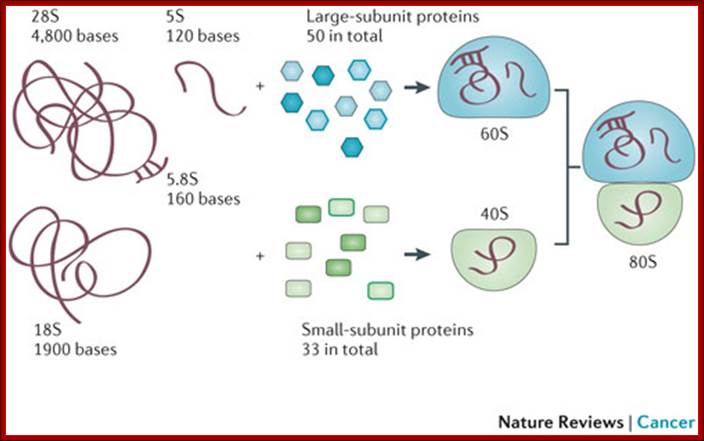
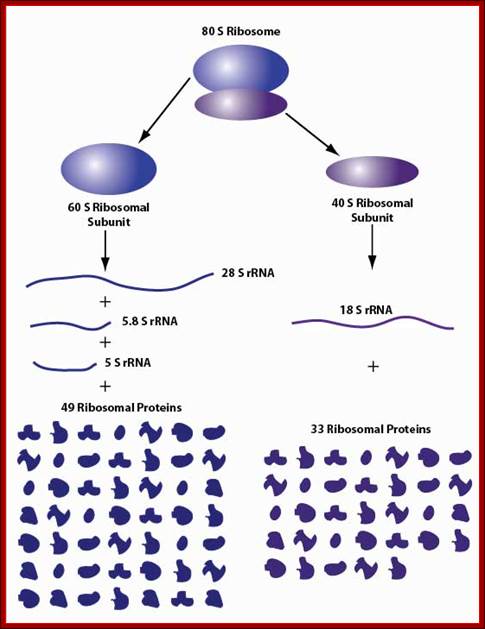
80s ribosomal components: 60s and 40s subunits,; rRNA sizes and associated proteins; 49 and 33 proteins; Ribosome biogenesis begins in the nucleolus with the co-transcription of RNAs encoding pre-ribosomal proteins and precursor polycistronic RNAs that are subsequently processed into three of the ribosomal RNAs (28S,18S and 5.8S). The ribosomal RNA genes are present in several hundred tightly clustered copies in the genome, unlike the scattered, but coordinately expressed, ribosomal protein genes. Intermediate assembled subunits are rapidly transported to the cytoplasm where final processing and incorporation of additional proteins to generate the mature 40S and 60S subunits is completed (see the figure). Approximately 200 different components have been found to function in the mammalian ribosome biogenesis pathway.
Mammalian ribosome biogenesis; Azra Raza & Naomi Galili; http://www.nature.com/ (Top Fig)
Ribosomal RNA gene expression and State of chromatin during expression
All eukaryotic cells irrespective of species, do exhibit rRNA synthesis all the time in the nucleolar region of the nucleus. In fact rDNA is considered as the ‘king of housekeeping genes’. Nucleolar size changes, when cells are activated indicating increase in the rRNA transcription. In the case of IBA induced root initiation in Phaseolus vulgaris, the size of the nucleolus is in pericyclic cells, so large it occupies ¾ of the nuclear volume. This shows the requirement of rRNA for pericyclic cells not others. Requirement of rRNA for any cell is always high and so it requires the transcription of rRNA all the time. To supply such high quantity, rRNA genes in chromosomal secondary constriction region (NOR region), loop out as naked DNA consisting of 100 to 200 rRNA genes in tandem repeats, depending upon the species and the stage of development.
In humans, chromosome 13, 14, 15, 21 and 22 contain rRNA genes in a region called NOR or nucleolar organizer region. Yeast contains rRNA genes on chromosome XII, Maize on chromosome 9, Drosophila on chromosomes X and Y. In Chironema NOR is located on 2 and 3 chromosomes. Xenopus laevis contains NOR on chromosome 5. After telophase as the nuclear membrane reconstitutes, chromosomes relax and open; at this point of time rRNA coding DNA open out in the form of loops of various sizes and organize into nucleolus, where the DNA is freed from all chromatin proteins gets associated with transcription complex and ‘transcription factory’ goes on producing rRNAs in massive amounts. And one can visualize each of the rRNA genes that are engaged in transcription, on an open DNA. One can observe Christmas tree like transcripts arranged-from the nascent to old transcript. In this the chromosomal DNA is totally devoid of any histones, and the only proteins associated are RNAP I and its associated factors are found. This goes on 24 hrs a day and 365 days a year. This implies for massive transcription the DNA should be free for the transcription complex to operate.
Nucleolar Dominance:
In diploid animals, inherited from two different parental genomes, it has been found that nucleolar organizer genes express from both parental chromosomes. But in certain members including plants and animals, the nucleolar organizers express from only one parental type and the other completely repressed. It has semblance of X chromosome inactivation in females in mammals.
http://sites.bio.indiana.edu/

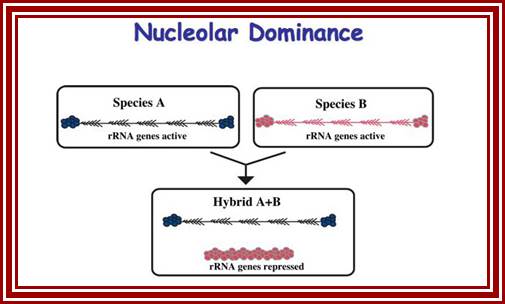
Nucleolar dominance; Pikard Lb-http://sites.bio.indiana.edu/
It is known that nucleolar loci exist in certain chromosomes and not in all. In plants chromosomes are inherited by the same parents, i.e. but pollen-male and egg-female are contributors. It so happens, in some the nucleolar DNA that opens up for transcription is either from one parent or the other. This process is very unusual. So one set of rRNA genes are kept silent and the other is expressed; which is similar to X chromosome inactivation in humans.
Nucleolar dominance is a common phenomenon in hybrid plants and it has also been studied in hybrid frogs (Xenopus), hybrid flies (Drosophila), and mammalian somatic cell hybrids. Nucleolar dominance was among the first epigenetic phenomena to be described that involves reversible gene-silencing on a scale perhaps second only to the inactivation of one X chromosome in somatic cells of female mammals.
In hybrids, genes inherited from both parents are typically expressed, producing intermediate phenotypes for characters such as flower or leaf morphology. This is true for Arabidopsis thaliana (A.t.), Arabidopsis arenosa (A.a) and their hybrid, A. suecica (A.s.). However, some genes are expressed from the chromosomes inherited from only one parent. An example is nucleolar dominance an epigenetic phenomenon in hybrids which describes the formation of a nucleolus (or nucleoli) on the chromosomes inherited from only one of the progenitors, regardless of whether that progenitor was the maternal or the paternal parent. Nucleolar dominance occurs in both the plant and animal kingdoms and is due to the expression of only one parental set of rRNA genes.
In 1997 plant mol. biologists found that nucleolar dominance in Brassica hybrids or A. suecica can be reversed by chemical inhibitors of DNA methylation or histone deacetylation which led to the realization that nucleolar dominance is due to the selective silencing of one set of rRNA genes rather than the selective activation of the other. Because rRNA genes are clustered by the hundreds, spanning millions of base pairs of chromosomal DNA, nucleolar dominance is one of the most extensive gene silencing phenomena known.
Further study has shown that rRNA gene silencing involves concerted changes in DNA methylation and histone modification and authors have proposed a model whereby DNA and histone modifications are each upstream of one another in a self-reinforcing, circular pathway. Changes in DNA methylation, histone acetylation and histone methylation are critical to the on-off switch mechanism that controls the number of active rRNA genes, both in hybrids displaying nucleolar dominance and in non-hybrids that regulate the effective dosage of their rRNA genes in response to the physiological needs of the cell.
The above authors have shown that rRNA gene silencing involves concerted changes in both DNA methylation and histone modification and the above authors have proposed a model whereby DNA and histone modifications are each upstream of one another in a self-reinforcing, circular pathway. Recent evidence indicates that siRNA-directed de novo cytosine methylation is involved in nucleolar dominance and may help explain how one set of rRNA genes is singled out for silencing.
RNA polymerases IV and V (Pol IV and Pol V) are plant-specific polymerases required for small interfering RNA (siRNA)-directed DNA methylation and silencing of transgenes, transposable elements, pericentromeric repeats and essential rRNA genes. These structurally and functionally distinct polymerases act in a pathway that also includes DICER-LIKE3 (DCL3), ARGONAUTE4 (AGO4) and RNA-DEPENDENT RNA POLYMERASE2 (RDR2).
In 2008, Pickard discovered evidence that biogenesis of 24 nt siRNAs is required for nucleolar dominance. Knocking down RNA-Dependent RNA Polymerase 2 (RDR2) or DICER-LIKE 3 (DCL3), which are required for 24 nt siRNA production, is sufficient to turn on the normally silenced A. thaliana rRNA genes in A. suecica. A 24 nt siRNAs direct the de novo methylation of corresponding DNA sequences, consistent with our finding that DRM2 is also required for nucleolar dominance. Because differential base pairing of siRNAs with A.thalliana versus A. arenosa rRNA gene sequences has the potential to explain the choice mechanism in nucleolar dominance, we are eager to understand the role of siRNAs in rRNA gene silencing. Pikaard lab research, Indiana University, Bloomingtom,IN.

Arabidopsis HDA6 is a histone deacetylase, previously implicated in the transcriptional silencing of transposable elements and transgenes, that is also required for nucleolar dominance, the uniparental silencing of ribosomal RNA genes in a genetic hybrid. Shown here is an immunofluorescence image of a leaf cell nucleus expressing Flag-tagged HDA6, stained with antibodies against HDAC6-Flag (red). Nuclear chromatin is counterstained with DAPI (blue). HDA6 is concentrated at multiple foci, the most prominent of which is the DAPI-negative nucleolus (top center), consistent with a role for HDAC6 in nucleolar dominance. Cover image courtesy of Olga Pontes. (For details, see Earley et al., p. 1283.).

Kinetics of rRNA processing proteins during nucleolar reconstruction. During anaphase, proteins of early (fibrillarin, green spots) and late (Nop52 and B23, red spots) rRNA processing machineries colocalize at the periphery of chromosomes (dark fiber). At the beginning of telophase, these proteins regroup in PNBs; early and late rRNA processing proteins pass through the same PNBs. Recruitment of rRNA processing proteins in the nucleolus then occurs according to differential sorting from the same PNBs of early (fibrillarin into dense fibrillar component; DFC) and late (Nop52 and B23 into granular component; GC) processing proteins (see Results). As a consequence, at the beginning of telophase, PNBs contain all the rRNA processing proteins. In contrast, after recruitment of the early rRNA processing proteins by the incipient nucleolus, PNBs only retain the late rRNA processing proteins. Interactions detected between proteins of the same rRNA processing machinery (visualized by bidirectional arrows between Nop52 and B23) in both PNBs and nucleolus suggest that PNBs are preassembly platforms for rRNA processing complexes; http://www.molbiolcell.org/
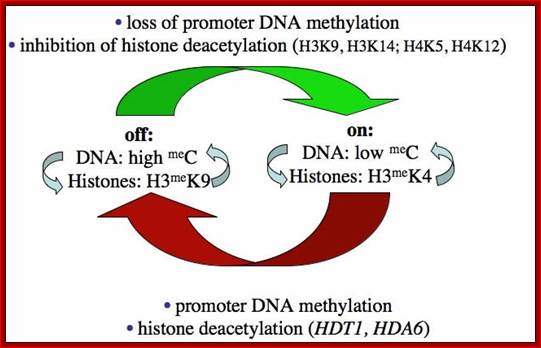
Chromatin modifications affecting ribosomal RNA gene activity. The model shows the chromatin modifications, and the names of corresponding Arabidopsischromatin-modifying activities (in bold), that help specify the active or silenced states of ribosomal RNA genes (rRNA genes) in Arabidopsis suecica hybrids displaying nucleolar dominance.; Craig S.Pickaard, http://www.hhmi.org/
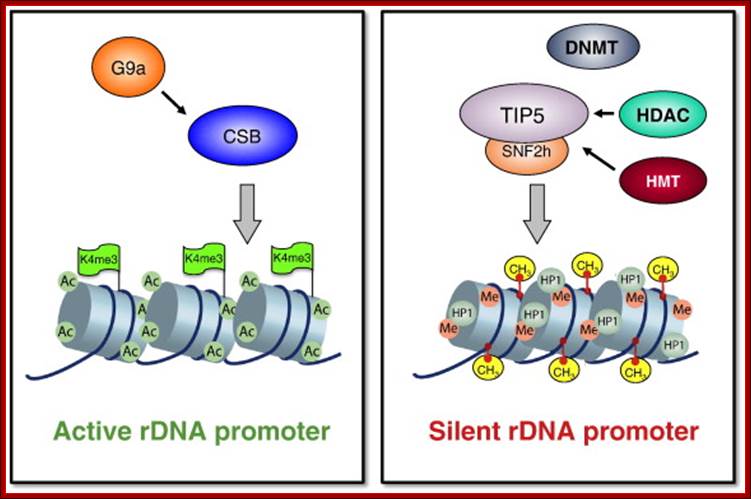
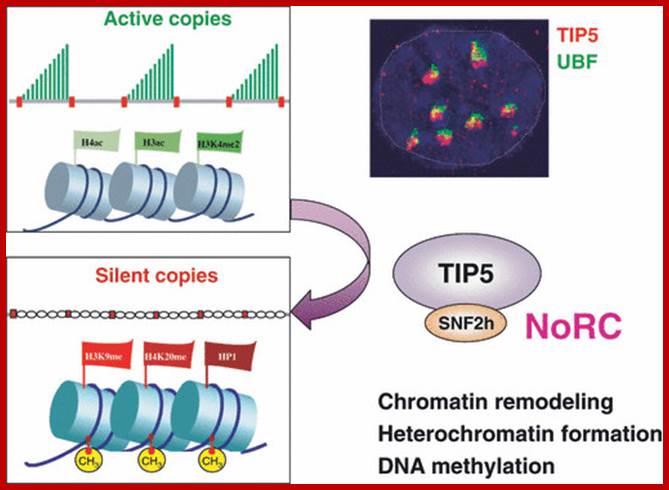
One of the genomic Nucleolar domain induces methylation at CpG island of the other promoter region of the DNA thus only one of the Nucleolar genes are expressed. NoRC triggers the establishment of the silent, heterochromatic state of rRNA genes. Potentially active rRNA genes exhibit an ‘open’ chromatin structure, are associated with Pol I and nascent pre-rRNA (green lines), and are characterized by DNA hypomethylation, acetylation of histone H4 (H4ac), and dimethylation of histone H3 Lys4 (H3K4me2). Epigenetically silenced rRNA genes are demarcated by histone H4 hypoacetylation, methylation of H3K9 (H3K9me) and histone H4 Lys20 (H4K20me), association with heterochromatin protein 1 (HP1) and CpG methylation (CH3). Methylation prevents UBF binding and impairs transcription complex formation. The silent state of rRNA genes is mediated by the NoRC, a complex comprising SNF2h and TIP5, which interacts with pRNA and histone-modifying enzymes. A deconvolution micrograph of interphase nuclei in U2OS cells, showing the nucleolar localization of TIP5 (red) and UBF (green) combined with 4′,6-diamidino-2-phenylindole-stained chromatin, is shown at the right. https://www.researchgate.net
The Pikaard lab has shown that Pol IV is required for the production of small interfering RNAs (abbreviated as siRNAs) that specify the silencing of matching DNA sequences, whereas Pol V makes longer RNAs that are thought to pair with the siRNAs at the affected chromosomal sites. Plant polymerases IV and V are special forms of Polymerase II ; ‘Chips off the old block’ By Tony Fitzpatrick; http://www.hhmi.org/
RNA-directed Transcriptional Gene Silencing in Mammals;Elizabeth H. Bayne, Robin C. Allshire
RNAi mediated gene silencing is achieved by DNA methylation and chromatin modification. Although further investigation is required to determine how widespread RNA-directed DNA methylation is in mammals, the findings raise the possibility that this pathway, far from being merely a curiosity of plant systems, is a conserved mechanism for control of gene expression.

Models for siRNA-directed chromatin modification: DNA (DMT) or histone (HMT) methyltransferase activity is thought to be recruited to target loci by a RITS-like complex that includes an Argonauts protein and an siRNA. Two potential mechanisms for target recognition are: (a) siRNA binding to target DNA; or (b) siRNA binding to nascent transcripts produced from target DNA by RNA polymerase II (pol II). Elizabeth H. Bayne
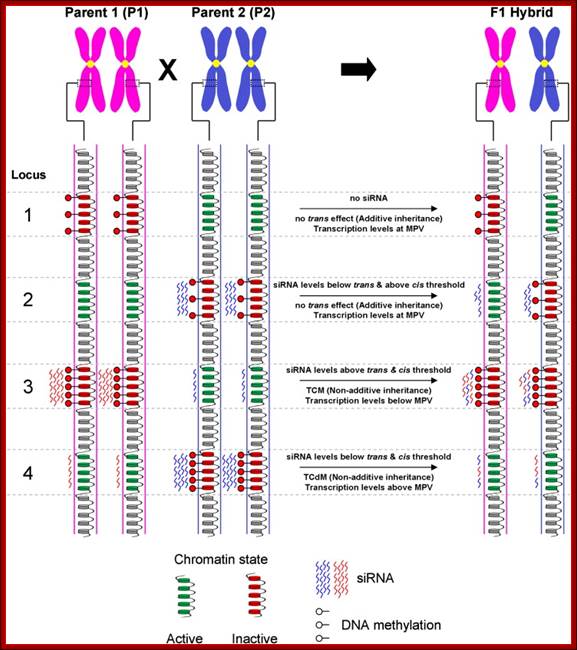
Model for inheritance of locus-specific siRNA and DNA methylation levels in the F1 hybrid: This is an example how chromatin can be made silent. A schematic representation of four hypothetical loci and associated epi-alleles with variations in chromatin states, DNA methylation and/or siRNA levels between parental lines [Note: (i) parental lines are homozygous for their respective alleles (ii) only one chromatid from each chromosome is depicted (iii) the specific contexts of DNA methylation not differentiated]. The transcriptional activity of each allele is dictated through the associated chromatin state which is influenced by siRNA and DNA methylation. In turn, the chromatin state can influence the levels of siRNA required to establish DNA methylation in the region. Several possible interactions may account for the hybrid epigenome differing from the expected average of the two parental lines (i.e. mid-parent value—MPV). Locus 1—Additive inheritance of the parental alleles resulting in transcription levels being at MPV. The presence of siRNA is a requirement for trans effects to occur. The lack of siRNA associated with the hypermethylated P1 allele results in additive inheritance in the hybrid. Locus 2—In this example, siRNA are associated with the methylation of the P2 allele. In the hybrids the P2 derived siRNA molecules associate across both alleles resulting in levels sufficient to retain methylation at the P2 allele but not enough to exceed the threshold set by the active chromatin state of the P1 allele. Therefore, the parental alleles retain their native methylation states resulting in additive inheritance and MPV transcription levels. Locus 3—The P1 derived siRNA molecules associate with both alleles, remaining above threshold levels to not only maintain methylation at the P1 allele, but to also establish de novo methylation of the hypo methylated P2 allele. This process of Trans Chromosomal Methylation (TCM) results in both parental alleles being methylated and silenced in the hybrid leading to below MPV transcription levels. In addition, the MPV siRNA levels are now being equally contributed by both parental alleles possibly as a consequence of the TCM process and the reinforcing interaction that exists between DNA methylation and siRNA production. Locus 4—In the hybrid, the P2 derived siRNA initially become associated across both parental alleles. This co-association results in siRNA levels at each allele falling below the required threshold to establish or even maintain DNA methylation causing the hypo methylation of the P2 allele. The loss of DNA methylation may also interrupt the process maintaining siRNA levels resulting in the subsequent loss of siRNAs and further reduction of DNA methylation. This process of Trans Chromosomal de-Methylation (TCdM) results in both parental alleles being active in the hybrid, leading to above MPV transcription levels; Michael Groszmanna, Ian K. Greaves et al.
Role of Arabidopsis ARGONAUTE4 in RNA-Directed DNA Methylation Triggered by Inverted Repeats; Daniel Zilberman1, 5,Xiaofeng Cao3, Lisa K Johansen4, Zhixin Xie4, James C Carrington4,Steven E Jacobsen.
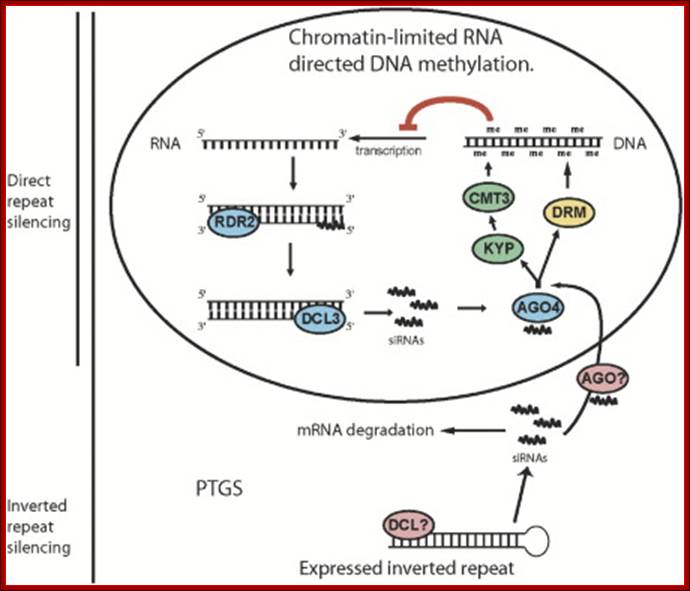
Model for RNA-Directed DNA Methylation in Arabidopsis: Diagram inside of the circle represents a hypothesized chromatin-limited siRNA silencing pathway in which siRNAs direct DNA methylation. siRNAs made by RDR2 and DCL3, together with AGO4, target DNA methylation through the action of KYP, CMT3, and DRM proteins. The bottom pathway represents PTGS, in which double-stranded RNA is processed into siRNAs by an unknown DICER protein. These siRNAs are used to degrade target mRNA and contribute to RNA-directed DNA methylation independently of AGO4, possibly through another AGO protein. We propose that RNA-directed DNA methylation driven by inverted repeats utilizes both pathways while direct repeat silencing is limited to the nuclear AGO4-dependent pathway. Daniel Zilberman et al
The high number of genes does only in part reflect the cellular demand for rRNAs, as only a fraction of these repeats is used for rRNA synthesis at any given time. In metabolically active human or mouse cells, approximately half of the 400 rRNA gene copies are transcriptionally active and the other half is silent. Once initiated, RNA polymerase 1 is capable of elongating through reconstituted chromatin without apparent displacement of the nucleosomes. In some plants the SC /NOR from one of the parents remains inactive just like human X chromosomes.
Another silencing locus in S. cerevisiae is the highly repetitive rDNA locus on chromosome XII. The rDNA locus encodes the ribosomal RNAs, which are not translated into proteins, but establish the ribosome together with ribosomal proteins. One rDNA unit consists of ~9.1 kb, which is tandemly repeated in 100-200 copies per cell. Silencing at this locus is thought to regulate the access of RNA polymerase I, but prevents RNA polymerase II from transcription (Shou, et al., 2001).
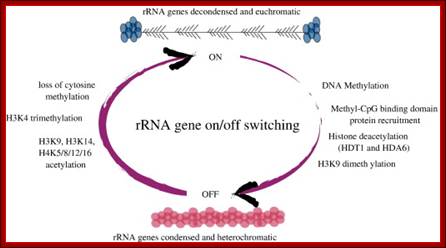
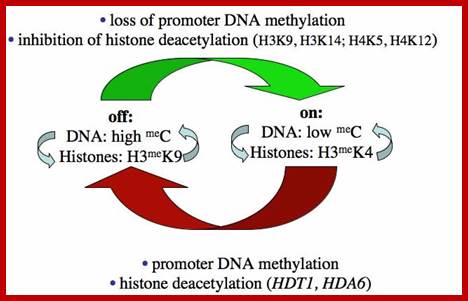
http://www.indiana.edu
Tandem repeats of rRNA genes decondensed and active in transcription; On and Off states.
At the rDNA locus, Sir2 (Silence information regulator) assembles together with Net1, Cdc14, Nan1 and PolI into the so-called RENT (regulator of nucleolar silencing and telophase exit) complex. In sir2Δ cells, the rDNA silencing is decreased and the chromatin structure is less compact whereas the recombination rate is increased). Thus, Sir2 improves rDNA silencing via its deacetylation activity (Smith and Boeke (1997) and Gottlieb and Esposito (1989).
The structural core component of the RENT (regulator of nucleolar silencing and telophase exit) complex is Net1, which recruits the other RENT-proteins to the rDNA locus. Net1 binds within the RENT complex to PolI, the RNA polymerase I, and stimulates its enzymatic activity. Whether Net1 directly binds to DNA is still unknown, but it is possible that other still unknown proteins contribute to rDNA silencing. Interestingly, when NET1 is over expressed, it is also associated with the HMR-E silencer, whereas net1-1 acts indirectly in HMR silencing by releasing Sir2 from the nucleolus (Kasulke, et al., 2002), (Shou, et al., 2001). Another component of RENT is NAD+-dependent histone deacetylase Sir2. Deacetylation leads to methylation. All these events lead to the binding of HP proteins and condensation of chromatin and silencing of chromatin.
Organization of rRNA genes:
In eukaryotes rRNA genes are mostly located in Secondary constriction (Sc) or nucleolar organizer (NoR) regions located in specific chromosomes and in specific positions. For example in human cells Scs are located on chromosomes 13, 14, 15, 20 and 21 just behind satellite structures. In yeast, S. cerevisiae, NOR is located on chromosome XII. The number rRNA genes in each of the NoR vary from 50 to 100 or more, all together number can be 150 to 500. Almost all rRNA genes are organized as tandem repeats with in between ETS (external non-transcribed spacers. Yet the ETS do contain few regulator elements in upstream of the rRNA operon.
II---rGene--II--rGene--II-rGene-II--rGene--I-ene(n)---II-
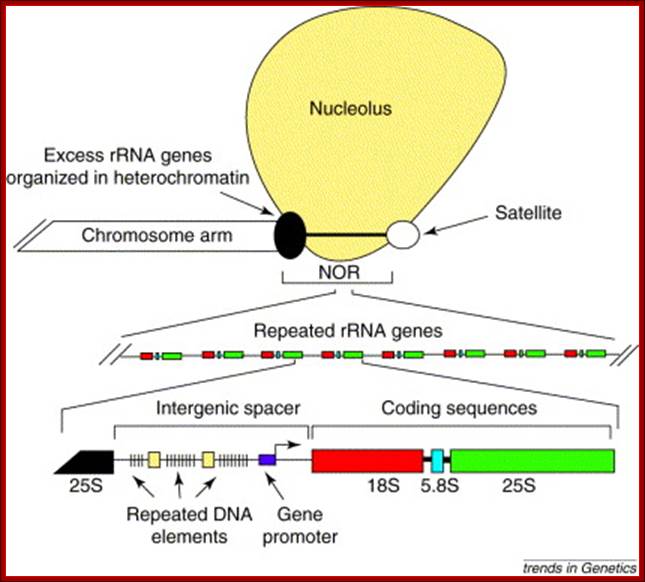
Organization of a typical nucleolus organizer region: Nucleolus orgainizer regions (NORs) are head-to-tail arrays of genes encoding the precursor of the three largest ribosomal RNAs (18S, 5.8S and 25S in plants). NORs include active rRNA genes, which give rise to secondary constrictions of metaphase chromosomes, and silent rRNA genes, which are often highly compacted in dense heterochromatin. At metaphase, a proteinaceous remnant of the nucleolus often remains associated with the secondary constriction. Each rRNA gene at a NOR is nearly identical in sequence, although variation in size due to differences in the number of repeated DNA elements in the intergenic spacer region is common. Craig S Pikaard
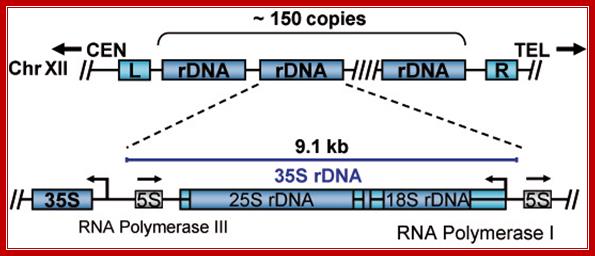
Yeast (S.cerviciae) rRNA genes:
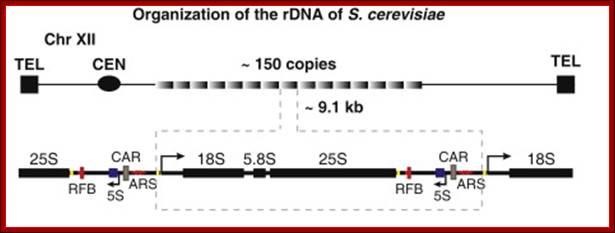
Schematic representation of the yeast rDNA locus
The position of the rDNA repeat cluster on chromosome XII including the left (L) and the right (R) flanking regions with respect to centromere (CEN) and telomere (TEL) is shown. Each rDNA repeat consists of the Pol I transcribed 35S rRNA gene (precursor for the 18S, 5.8S, and 25S rRNAs) and the RNA Polymerase III.
,
Position of rRNA 150 gene clusters just behind Telomere, one rRNA gene expanded
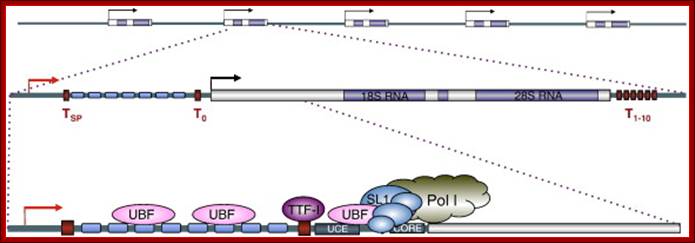
Part of the rRNA gene clusters. A single rRNA gene with assembled transcriptional apparatus in the upstream of the gene for transcription
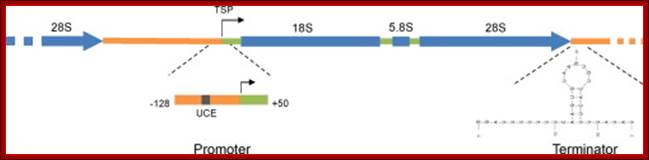
An eukaryotic rRNA gene structural feature.
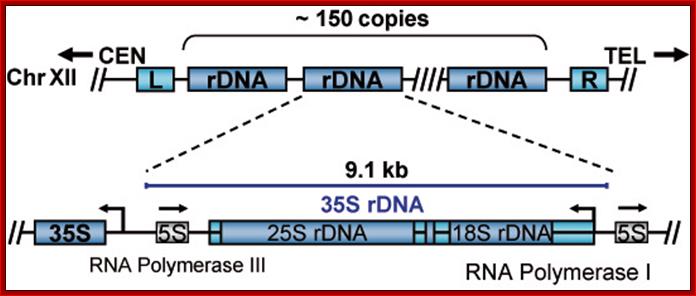
In yeast NOR is located on XIIth chromosome. The structure of the S. cerevisiae rDNA and the model of rRNA gene transcription by Pol I: It also contains 5S RNA genes on either side of rRNA operons. 5S RA genes are distinct and transcribed by RNApol III. Arrows mark transcription start and direction. House of the Ribosome; rDNA Chromatin –Joachim Griesenbeck; \http://www.biologie.uni-regensburg.de
[Enh-à-18s-5.8s-25s----[<--5s ] >-----ETS 18s>—5.8s—25s---[-5s-] >--Enh]
A single copy of rDNA repeats is shown. The 9.1 kb unit consists of the 35S rRNA (25S and 18S) coding region, the 5S rRNA gene is located in between two 35S RNA genes and it is transcribed independently by RNAPol III.

1. The NTS has several domains involved in transcriptional activation; in between two such 35s segment one finds 5s rna segment which is transcribed by RNAPIII in oposite direction. The intergenic rRNA gene spacer of the yeast S. cerevisiae, (A) One unit of the rRNA gene repeat consists of 9.1 kb of DNA. It contains the following elements: the 35S-rRNA gene transcribed by RNAP-I (white box; 35S); a 5S-rRNA gene transcribed by RNAP-III (black box; 5S); a ribosomal origin of replication (gray box; ARS); an enhancer (white box; E); and two nontranscribed spacers between the 35S and 5S genes (NTS1 and NTS2). Relevant restriction sites and fragments (AvaII, ClaI, NheI, NdeI, 3.9 kb, and 3.4 kb), the probe used for indirect end labeling from the NdeI site (black bar), and the primer used for high resolution foot printing (fat horizontal arrow) are indicated. (B) Schematic illustration of the promoter elements and transcription factors: upstream element (UE), core element (core), UAF containing Rrn5, Rrn9, Rrn10, Uaf30, histone H3, and histone H4, the CF containing Rrn6, Rrn7, Rrn11, the TATA binding protein (TBP), and the RNA-polymerase I (RNAP-I) with the associated Rrn3. Molecular and Cell Biology; Andreas Meier† and Fritz Thoma; http://mcb.asm.org/
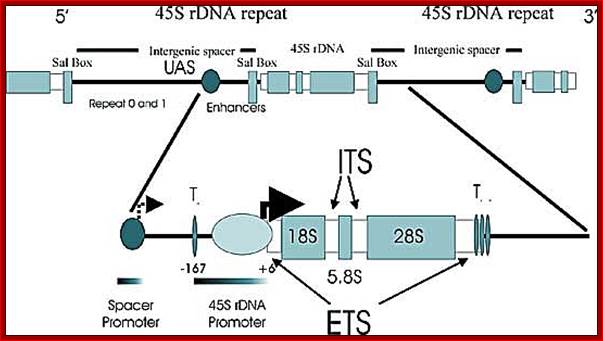
Top Fig; In actively dividing cells rRNA synthesis by RNAPI is responsible for the synthesis of 70% of the total transcription in the yeast cell. Bottom fig; Between each of rRNA genes there is an intergenic spacer and it is used for Transcriptional initiation and the upstream is used as enhancer elements. The gene has external transcription spacers and inter transcriptional spacer; http://www.biologie.uni-regensburg.de/
rGene = rRNA gene,
UAS = Upstream activator sequences,
ETS =External non transcribed sequence,
ITS = Internal transcribed sequence,
IGS = Intergenic spacer
T ter = transcriptional terminator region.
Sal Box-SalI restriction site.
Ribosomal pre-RNAs are transcribed in the nucleolus and then they are processed. There are specialized sn RNAs (U3Sno RNA and SnoRnps), Sno RNAs and Sno-Rnps to perform processing.
Processing of pre-rRNA:
Pre rRNA is long and it is processed in terms removing certain portion of internal spacers and certain specific nucleotides are modified i.e. methylation or pseudouridinylation for they perform specific functions. Slicing is assisted by U3 sn/snoRNA and modifications are aided by snoRNAs and specific enzymes. The process of methylation and pseudo-uridine synthesis takes place in the Nucleolus just before the cleavage of precursor rRNA or during the cleavage process. However it is assumed that methylation and uridine modifications at specific sites provide the land mark for pre-rRNA processing (which includes cutting, trimming and nucleotide modifications).
Cajal Bodies:
New clues to the function of the Cajal body, Maria Carmo-Fonseca
Cajal bodies (CBs) are small spherical structure found in the nucleus and they are not bound by any unit membranes. They contain RNA and proteins complexed. A new family of small RNAs is found to be localized specifically in Cajal bodies and they are involved in the maturation of spliceosomal snRNAs (Darzacq et al., 2002). Bertrand and co-workers showed that the final processing steps involved in the biogenesis of U3 sn (o) RNA take place in Cajal bodies. Santiago Ramony Cajal discovered these structures in 1903, that is more than 100 years ago. Cajal bodies (CBs) are spherical sub-organelles of 0.3-1.0 µm in diameter found in the nucleus of proliferative cells like embryonic cells and tumor cells, or metabolically active cells like neurons. CBs have been implicated in RNA-related metabolic processes such as snRNPs biogenesis, maturation and recycling, histone mRNA processing and telomere maintenance. CBs assemble RNAs which are used by telomerase to add nucleotides to the ends of telomeres (WIKI). CBs contain several small RNAs called ‘sca’ RNA. These are involved in post-transcription processing of snRNAs. The small Cajal body-specific RNAs (scaRNAs) are predicted or have already been demonstrated to function as guide RNAs in site-specific synthesis of 2'-O-ribose-methylated nucleotides and psuedouridines in the RNA polymerase II-transcribed U1, U2, U4 and U5 spliceosomal small nuclear RNAs (snRNAs).
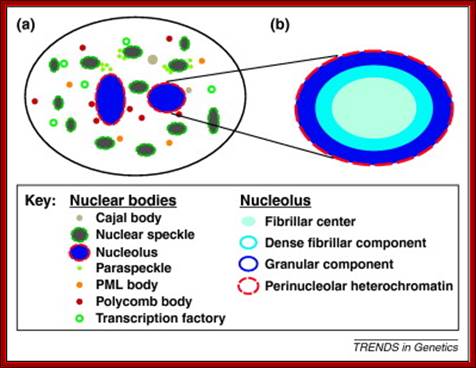

Cajal Bodies in Nuclear sap.
|
Subnuclear structure sizes |
||
|
Structure name |
Structure diameter |
|
|
Cajal bodies |
0.2–2.0 µm |
|
|
PIKA |
5 µm |
|
|
PML bodies |
0.2–1.0 µm |
|
|
Paraspeckles |
0.2–1.0 µm |
|
|
Speckles |
20–25 nm |
|
PIKA=Polymorphic interphase karyosomal associated, PML = Promyelocytic leukemia bodies, Paraspeckles = Part of Nuclear speckles.

This excellent picture shows the sites called Cajal Bodies where the Sno RNAs-RNPs mediated rRNA modifications of a variety of sn RNAs take place.
Nucleus contains many specialized structures, among them Cajal bodies are very prominent and they contain coiled-coiled proteins called Coilins. These bodies are visible when fluorescence staining is used, and they also contain RNAP I, II and III, some nuclear transcripts, TFII F and TF-IIS. It also contains SR proteins and splicing factors and Poly (A) adenylation factors, snurposomes and few other RNA processing enzymes. Many of cap containing sn RNAs move out of the nucleus into cytoplasm and they are further methylated (Tri) and imported back as 2’2’7-CH3 methylated snRNAs and land up in Cajal bodies, where some of them are modified similar to rRNAs. Sno RNA U85 has both H/ACA and C/D domains. The Sca sno-RNAs also modify some mRNAs. One of the Sn RNA first detected within Cajal bodies was U6 URNA. This snRNA is not coded by RNAP II but by RNAP III.
This U6 snRNA contains three pseudouridines and eight 2’O-CH3 groups. It is not just U6 RNA, there are other Sn RNAs such as U1, U2, U4, U5 are also modified and all in all they contain 7’G-2’O-2’OCH3 groups and 24 pseudouridine groups. Even few mRNAs such as serotonin (neuronal) are methylated and pseudo uridinylated. Some of the sno RNAs even modify tRNA by adding CH3 groups to bases and converting uridine to pseudouridine. The sno RNAs responsible to perform such reactions are called Sca RNAs. One of the sno RNA called U85 has the sequences for 2-O methylation C/D box and H/ACA box for pseudouridine synthesis such sno RNAs are called Sca RNAs.
New Types of Guide RNAs, Cellular Locations, and Substrates in Eukaryotes:
In a surprising development, new candidate human guide RNAs have been discovered to reside exclusively in Cajal (coiled) bodies. The small Cajal body-specific RNAs, called scaRNAs, have the hallmark features of the archetypical snoRNAs, except that some have an unusual arrangement and content of guide motifs. Among the current set of scaRNAs are: species with both Nm and Ψ modification motifs and species that resemble the classically defined guide snoRNAs. Guide sequences have been identified thus far in the scaRNAs for known modifications in pol II-transcribed snRNAs (U1, U2, U4, and U5).
Adding to the excitement, new results with yeast have identified novel structures called nucleolar bodies (NBs), which have overlapping functions with Cajal bodies. Newly synthesized U3 and U14 snoRNAs, and C/D and H/ACA snoRNA/RNP proteins have been detected in the NBs. Strong evidence that both snoRNAs and snRNAs undergo maturation events in nucleolar bodies comes from the discovery that an enzyme, Tgs1p, that hypermethylates the 5′-cap structures of a subset of snoRNAs and the pol II snRNAs appears to be localized to this structure. Consistent with parallel functions of CBs and NBs, the vertebrate ortholog of Tgs1p occurs in the Cajal bodies, suggesting that cap formation of snoRNAs takes place in this nuclear compartment. (Note that most vertebrate snoRNAs undergo 5′-end processing and lack caps.) The parallel is not complete, however, as hypermethylation of pol II snRNAs is known to occur in the cytoplasm and Tgs1 protein localizes to the cytoplasm as well as the CBs.

Guided modification appears to occur in vertebrate Cajal bodies.
New snoRNAs enter the CBs before localizing to the nucleolus (NO); however, a subset is retained there. The small Cajal body-specific RNAs (scaRNAs) are complementary to pol II-derived snRNAs, which are also found in CBs. Modification of yeast snoRNAs and snRNAs may occur in yeast NBs, which share properties with Cajal bodies.

(A)
Processing of spliceosomal
U snRNAs. The U1, U2, U4 and U5 spliceosomal snRNAs are synthesized in the
nucleoplasm by RNA polymerase II as immature precursors containing a
5'-terminal 7-mono-methylguanosine cap and extra nucleotides at the 3' end (for
simplicity, only the U5 snRNA is depicted). These RNAs are rapidly exported to
the cytoplasm, where they bind to Sm proteins. The association of Sm proteins
allows subsequent hyper-methylation of the 5'-cap and 3'-end maturation of the
snRNA. The newly assembled snRNPs (small nuclear ribonucleoprotein particles)
are then transported back into the nucleus, where the snRNAs further undergo
2'-O-ribose methylation (Me) and pseudouridine formation (![]() ). The new data reported by Darzacq et al. (2002) suggest
that these modifications are guided by the U85, U87, U88, U89, U90, U91 and U92
small RNAs located in the Cajal body (scaRNAs). Modified nucleotides are
confined to the functionally important regions of snRNAs. (B) Processing of the
U3 snoRNA. The U3 snoRNA is synthesized in the nucleoplasm by RNA polymerase II
as a 3'-extended precursor with a mono-methylated 5' cap. Cap tri-methylation
is catalysed by the methyl-transferase Tgs1/PIMT, which localizes to the Cajal
body as shown by Verheggen et al. (2002). Following 3'-end processing,
which also takes place within the Cajal body, the U3 RNA binds fibrillarin
(Fib) and other core snoRNP proteins. The mature U3 snoRNPs leave the Cajal
body to accumulate in the nucleolus.
). The new data reported by Darzacq et al. (2002) suggest
that these modifications are guided by the U85, U87, U88, U89, U90, U91 and U92
small RNAs located in the Cajal body (scaRNAs). Modified nucleotides are
confined to the functionally important regions of snRNAs. (B) Processing of the
U3 snoRNA. The U3 snoRNA is synthesized in the nucleoplasm by RNA polymerase II
as a 3'-extended precursor with a mono-methylated 5' cap. Cap tri-methylation
is catalysed by the methyl-transferase Tgs1/PIMT, which localizes to the Cajal
body as shown by Verheggen et al. (2002). Following 3'-end processing,
which also takes place within the Cajal body, the U3 RNA binds fibrillarin
(Fib) and other core snoRNP proteins. The mature U3 snoRNPs leave the Cajal
body to accumulate in the nucleolus.
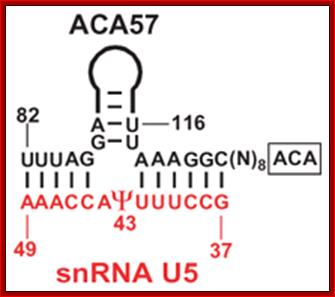
U3 Sn RNA: This is 210 ntds long, has 5’m7G cap structure, and no poly A tail. It is transcribed by RNAP III in plants but in lower eukaryotes it is transcribed by RNA pol II. The RNA gets associated with specific snRNPs. It cross-links with pre-rRNA. This U3 Sn RNA is involved in the first cleavage reactions from 5’ end.
Retention and 5' cap trimethylation of U3 snRNA in the nucleus- (MP Terns and JE Dahlberg):
It is shown here that maturation of m7G-capped precursors of U3 small nuclear RNA (snRNA) occurs by a previously unknown pathway. In contrast to the 5' m7G-capped precursors of other snRNAs, this RNA is not exported to the cytoplasm but is retained in the nuclei of Xenopus laevis oocytes, where it undergoes trimethylation of its 5' cap. The m7G caps of most snRNA precursors are trimethylated only after transport of the RNAs to the cytoplasm. The nuclear retention and maturation of this nucleolar RNA raises the possibility that other m7G-capped RNAs are also retained and modified in the nucleus (?).
The U3 snRNA (U3 snoRNA) molecules persist throughout mitosis in close association with the nucleolar remnants. U3 snRNA is present in the prenucleolar bodies (PNBs) and could participate in nucleologenesis in association with several nucleolar proteins such as nucleolin and fibrillarin. The interaction of U3 snRNP with the 5’external spacer of pre-RNA newly synthesized by active NORs is proposed to be the promoting event of nucleologenesis.
U3 sn RNA is transcribed by RNApIII and not by RNAPII. Initiation and termination of snRNA transcription occur by mechanisms different from those of other polymerase II transcripts. “This is the first in vivo demonstration that U3 snoRNA plays a role in rRNA processing”. It is postulated that U3 snRNP folds pre-rRNA into a conformation dictating correct cleavage at processing site at 3’.
Much of rDNA sequences are called nucleolus organizers, term these coiled body-associated sequences ‘coiled body organizers’ (CBORs). Human U3 genes are clustered on human chromosome 17p11.2, with evidence for large inverted duplications within the cluster (Liming Gao, Mark R. Frey and A. Gregory Matera). Thus all of the CBORs identified to date are composed of either tandemly repeated or tightly clustered genes. The evolutionary and cell biological consequences of this type of organization are discussed
Sno RNAs:
These RNA are small molecular weight RNAs of 120-300 nucleotides. More than 100 such sno RNAs have been identified. Most of them have specific sequences. Some of them are actually derived from the pre-mRNA intronic sequences. And some are coded for by specific genes transcribed by RNAPII. Pre-mRNA derived Introns are released by cis splicing and they contain lariat shape and the same are debranched. They soon get associated with specific proteins and reach Cajal Bodies, where the pre-rRNAs are subjected to nucleotide modifications.
There are two classes of Sno RNAs, one is called C/D Sno RNAs which perform sequence specific 2’O methylation; and the other is called H/ACA Sno RNAs, they perform site specific conversion of Uridines to Psuedouridines. The number of such RNAs varies greatly 50000 to 200000 per cell.
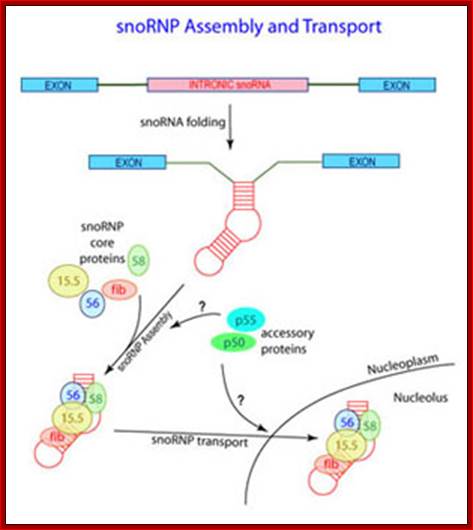
The cleavage and assembly of intronic RNAs into SnoRNA which soon get associated with their specific proteins as SnoRNPs; as a complex they act on precursor rRNA in sequence specific manner and modify the sugars or bases. Maxwell lab; http://biochem.ncsu.edu/
Some of the small mol.wt RNAs:
|
snO RNA |
size |
location |
Function |
|
Sno RNAs-C/D types (71) ? |
100-500, |
Nucleolus RNAP-II, derived from Introns |
Methylation of rRNA at ribose 2’OH positions (100 sites) |
|
Sno RNAs-H/ACA types (20)? |
55-250, |
Nucleolus, RNAP-I, derived from introns |
Pseudo-Uridinylation of rRNA (100 sites) |
|
U3 RNA |
~210 ntd |
Nucleolus-Cajal bodies |
First rRNA cleaving reactions |
There are 71 Sno RNAs C/D class in Yeast cells, associated with Fibrillarin proteins. There are 100 or more methylation sites in rRNAs.
There are more than 20 H/ACA type of Sno RNAs involved in converting Uridine residues at specific sites to pseudouridine (changing 1N bond to sugar to C5 to join sugar. They are associated with GAR-I proteins.
These Sno RNAs are also associated with their respective proteins called Sno-Rnps. Some SnoRNAs are spliced out introns or one can call them as intronic noncoding RNAs. And also some of them are introns of non-functional mRNAs and there are some transcribed by specific genes by RNAP II. Some Sno RNA genes are either monocistronic or polycistronic. The monocistronic or polycistronic snoRNAs are transcribed by RNA pol-II and they are processed and capped. U14 sno is polycistronic transcribed as independent gene.
---------aaRCCoTa-----TATA--à+1--
Overall there can be 200 or more types of Sno RNAs; they are transcribed by RNA Pol-II and RNA Pol III. Many of them are derived by excised introns.
Methylation:
Methylation in eukaryotic rRNA is mostly at 2’O of specific ribose sugar. This is performed by special small nucleolar RNAs called CD Sno RNAs and enzymes. They show secondary structure with two sequence boxes called C and D. The C box contains 5’---RUGAUGA--- and the D box has –CUGA towards the 3’ side of the same RNA. However, it has also further down not so perfect sequences called C’ and D’ boxes. There are other methylases which modify bases with methyl group.
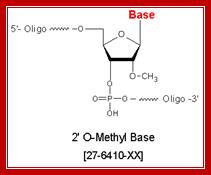
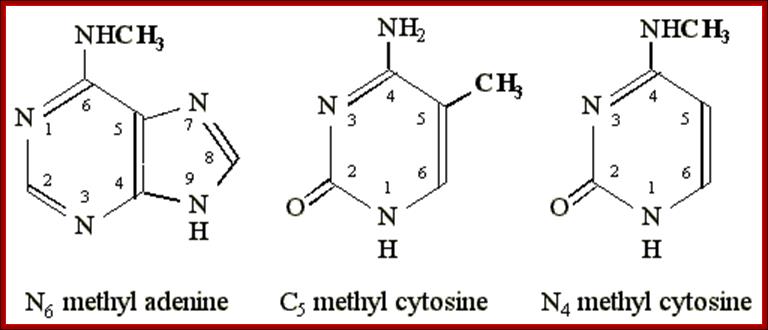
Molecular Biology and Biotecbhnology; Dr. Michael Blaber; http://www.mikeblaber.org/
These C/D RNAs are associated with specific proteins such as fibrillarins 65 and 68Kd, Nop-56Kd, Nop 58KD, Nop 50kd, Snu3p/15.5Kd and 7-2 MRP RNAse.
The H/ACA RNAs are also associated with GAR1p, CVF5p, NHp2p, NOP2p,10p, Dyskerin, Map7-2
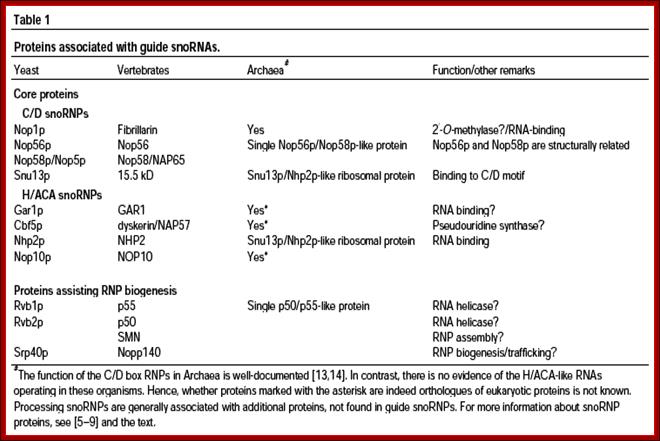
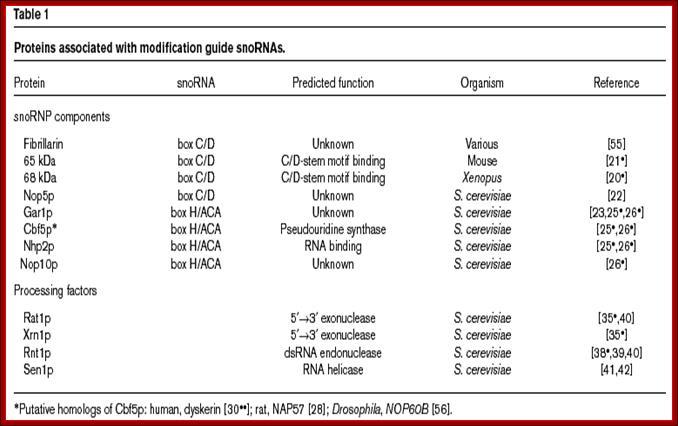
Sno RNA associated proteins-SnoRNPs:
|
C/D snoRNPs; methylation |
H/ACA-SnoRNps; pseudouridinylation |
|
Fibrillarin- 34Kd,68Kd |
GAR 1p |
|
NOP 58Kd, 50Kd,15.5 |
CVF5p |
|
Snu3p-15.5kd |
NHP2p |
|
7-2 snoRNase |
NOp2p,10p |
|
|
Dyskerin |
|
|
Map7-2 |
Mechanism:
As the rRNA is synthesized, the RNA undergoes structural conformation in sequence specific manner; this will provide specific sites for methylation at 2’OH of the ribose.
- The CD Sno RNA seeks specific sequences and base pairs to form C and D boxes. The rRNA using specific sequences base pairs with C/D boxes. The enzyme S-Adenosyl methyl transferase (SAM) targets the 5th nucleotide in rRNA from 5’end of the rRNA and methylates ribose at 2’OH group, similarly methylation takes place at D box also, but not always.
- There are ~100 or more sites at which rRNA gets methylated. The number and sites slightly vary from one system to the other. For any given system the positions of methylation is same. This facilitates the endonuclease site-specific cleavage, and also which provides skeletal work for folding and the assembly of riboproteins in hierarchical fashion.
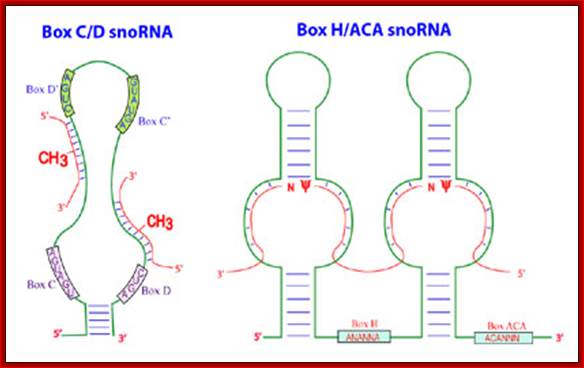
This is a line diagram showing the secondary structural configuration when the Sno RNA base pair with pre-rRNAs; the left diagram id CD Sno RNA and right figure is of A/HCA Sno RNA, The Maxwell Lab; http://biochem.ncsu.edu/
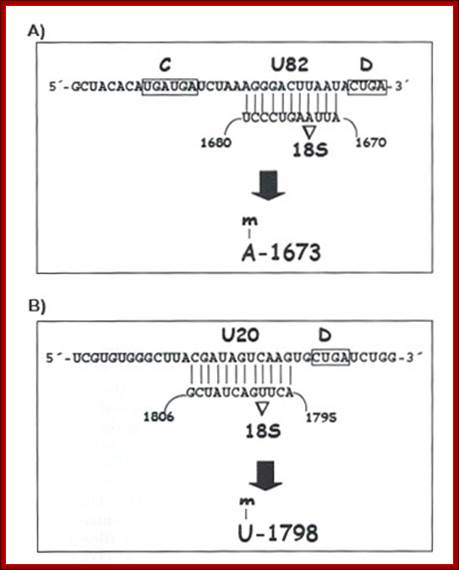
Sno RNAs base paired to pre-rRNA
A Table showing the number of sites modified in various systems:
|
Species |
Base-CH3 |
2’-O-CH3 |
Pseudo U |
Total |
|
E.coli |
22 |
4 |
10 |
~36 |
|
S.cerevisiae |
10 |
55 |
44 |
~112 |
|
S.solfotaricus |
~8 |
67 |
9 |
~88 |
|
X.laevis |
10 |
99 |
~98 |
~207 |
|
H.sapiens |
10 |
107 |
~95 |
~212 |
|
D.melanogaster |
|
|
|
~63 |
http://users.soe.ucsc.edu; [Bachellerie & Cavaille, 1998,Ofengand & Fournier, 1998,Noon et al., 1998].
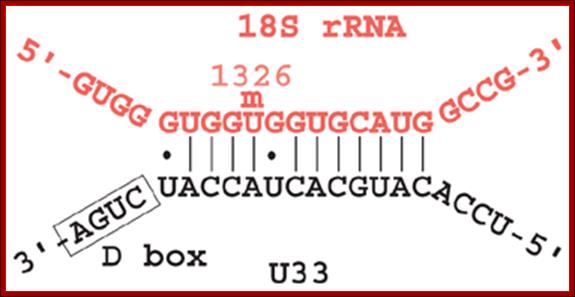
This diagram shows a specific Sno-RNP base pairing to specific pre-rRNA sequence for methylations at 2’OH group.
- Besides Sno RNAs the Sn U3 RNA is also involved in first few cleavage reactions. So also MRP RNase.
Conversion of Uridine to pseudouridine:
Another group of Sno RNAs are called H&ACA Sno RNAs. There are several such sno RNA species and they are involved in conversion of Uridine nucleotides at specific position into pseudouridine by enzymatic reactions. There are 43 pseudo Uridine in yeast rRNAs and 100 or so pseudouridine in vertebrates.
- These H/ACA Sno RNAs are associated with proteins such as GAR1p (Dyskerin), cbf5p (pseudouridine synthase), nhp2P, Nop1oP, Pop1-3 protein, MRP 7-2 RNase.
Mechanism:
This diagram shows an example of one of the Sno-RNAs involved in base pairing with the pre-rRNA for pseudo-uridinylation.

Pseudouridine
- The H&ACA Sno RNAs contain two stem loop structures separated by H-box (ANANNA) in between two stem loops and ACA box (ACANNN) on the right side of the second stem loop. Each of the stem loops contains two open loops one at the base and the other is terminal. These loops have specific sequences.
5’—stem-loop—H-Box—stem loop—ACA box—3’
- The rRNAs base pair at the basal open loop at specific positions, where two Uridines in each loop remain unpaired. These regions are recognized by a set of enzymes and they bind and convert the second Uridine in rRNA from the left (from the 3’end) into pseudouridine by cleaving the glycosidic bond at the N1 position and making a glycosidic bond to 5th position C of the Uridine. H and ACA Sno RNAs are associated withGar-1P proteins.
U8 snoRNA
- Most of these modifications assist the folding of the rRNA into secondary and tertiary structures, cleavage at specific positions and association of riboproteins into ribosome assembly
Orphan Sno RNAs:
There are many snoRNAs whose functions is yet to be determined, till then they remain or called orphan Sno RNA. Using sequences computer biologists are trying to find out the number and kind of Sno RNAs.
Biogenesis of Sno RNAs:
Sno RNAs are small molecular weight RNA whose length can be between 55 -500 or more. They are all single stranded but by complementary base pairing, among themselves assume characteristic secondary structures, but the association of snRNPs makes them active catalytic bodies.
They are derived from two different sources; one from the intronic sequences of pre-mRNAs and the others are transcribed by separate snoRNA genes. There are more than Sno RNAs in mammals and most of them are C/D SnoRNAs. The existence of separate genes has been established. They have their promoter and coding regions. They are transcribed RNAP II; sno RNAs have caps at 5’end and some are transcribed by RNAP III (they don’t have caps at 5’ends); they are processed at the 3’ end to final forms. The number of such RNAs varies greatly 50000 to 200000 per cell.

Genomic organization of snoRNA coding units: Schematic representation of the different types of genomic location of snoRNA genes. The snoRNA coding units endowed with independent promoters (top) and those located within introns (middle) are transcribed by RNA polymerase II. Frequently, neighbouring introns of the same host gene contains snoRNA coding units with a one-gene-per-intron distribution. In such cases, the snoRNA coding units have been considered as “intronic individual” even though several different snoRNAs can originate from the same precursor transcript. Independent genes are found in yeasts and plants-both mono or polycistronics, transcribed by RNAPII., some by RNAPIII,
The U3 is considered as SnoRNA. In vertebrates and invertebrate U3 transcribed by RAPII using OSE, in yeast by RNAPII it contains TATA box plus UPE. In plants and unicellular algae it is transcribed by RNAP II, and it contains UCE and TATA box.
The utilization of upstream Pol III-specific box A and box B to drive transcription of guide snoRNA genes has also been documented in the land plants A. thaliana and O. sativa, where a few guide snoRNAs are synthesized as dicistronic tRNA–snoRNA precursors, and in the nematode C. elegans. At least two Drosophila guide snoRNA genes are also independently transcribed by Pol III, possibly through the utilization of box B promoter elements. In conclusion, it appears as if autonomous snoRNA gene transcription was achieved rather opportunistically during evolution, through the flexible exploitation of different types of specialized promoters.

Processing of polycistronic sno RNAs and Intron derived sno RNAs; Terence I. Moy et al
Those derived from introns are debranched and assume secondary structure with open loops. While they are cut and released they get associated with specific proteins and they are then processed to final form.
The methylating snoRNPs have four common core proteins. The total number of proteins in a particle is not known nor is it known if all modifying snoRNPs are identical except for the RNA component. The core proteins in yeast (and humans) are: Snu13p (15.5 kDa), Nop56p (hNop56p), Nop58p (hNop58p), and Nop1p (fibrillarin). Snu13p binds to a characteristic stem-asymmetric loop-stem structure that includes the canonical C/D elements in the loop portion. Interestingly, Snu13p is also part of the U4 snRNP where it binds a similar, common RNA fold called the K-turn. Nop56p and Nop58p are related to each other and both interact with snoRNA.
Nop1p (fibrillarin) is generally accepted to be the 2′-O-methyltransferase
The four core proteins of the H/ACA snoRNPs differ from those in the C/D snoRNPs, and the same uncertainties exist about the numbers and types of proteins among the individual snoRNPs. The core proteins in yeast (and human) include: Cbf5p (Dyskerin), Gar1p (hGar1p), Nhp2p (hNhp2p), and Nop10p (hNop10p). Cbf5p is accepted to be the pseudouridine synthase.
Cutting and Trimming of pre-rRNAs:
- In eukaryotes also the processing is more or less similar. The number of genes, however, for rRNA found is in hundreds or more in number.
- All of them are located in specific chromosomes and at specific site called secondary constriction or nucleolar organizer (NOR); in Homo sapiens the secondary constrictions are found in five pairs of chromosomes-13, 14, 15, 21 and 22.
- As the nuclear membrane reforms at the end of Telophase, within the nucleus the rRNA gene clusters open out into naked DNA threads as loops, where each loop consists of several rRNA genes arranged in tandem repeats with non-transcribed spacer regions in between them.
- The number of such genes can be in several hundreds, localized in Secondary Constriction or Nucleolar Organizer region.
- Opened or looped out rRNA genes are again localized organize into a region called Nucleolus, separated from the rest of the nuclear karyolymph or nuclear sap by unknown or unstructured border.
- The rRNA genes transcribe and produce precursor rRNA transcripts, which eventually processed (modifications and cutting by specific enzymes) to final products.
- The transcripts associate with the imported riboproteins into large and small ribosomal subunits. The required riboproteins are produced in cytoplasm and then they are transported into the nucleolus across nuclear membrane pore-apparatus.
- Ribosomes associate with mRNA’s 5’ ends and transported into the cytoplasm by Ran-GTP export system.
- So, within the Nucleolus, one finds three regions called pars-Amorpha (liquid area), pars-Fibrosa (filamentous area containing RNA threads) and pars-Granulosa (granular ribosomes
- Outside the nucleolus, there are few nuclear bodies among them Cajal bodies are very important.
This is an excellent diagram of processing of an eukaryotic pre-rRNA by certain endonucleases and few exonucleases mentioned in the figure. The figure below also shows the process of cutting stepwise by specific nucleases. The diagram also shows alternate mode of processing of 27s RNA.
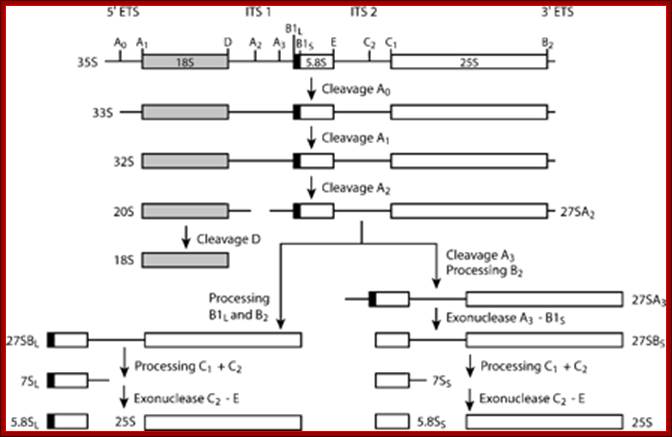
ETS= external transcribed spacers, ITS= internal transcribed spacers; Terence I. Moy et al; http://mic.sgmjournals.org/
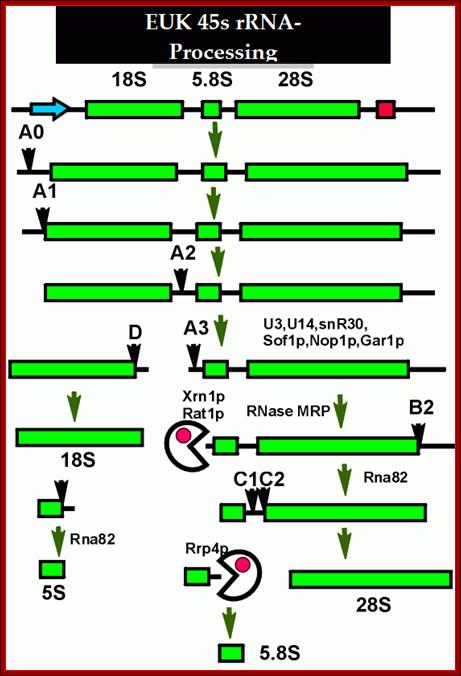
1. The major pre-rRNA processing pathways in S. cerevisiae. The 35S pre-rRNA is transcribed in the nucleolus and processed into the mature 18S, 5·8S and 25S rRNAs by a series of endonucleolytic and exonucleolytic steps. The 35S pre-rRNA is cleaved at site A0 to generate the 33S pre-rRNA and site A1 to generate the 32S pre-rRNA. The 32S pre-rRNA is then cleaved at A2 to produce the 20S and 27SA2 pre-rRNAs, thereby separating the RNAs destined for the small and large ribosomal subunits. Final maturation of the 20S precursor into the 18S rRNA occurs in the cytoplasm. The 27SA2 precursor is processed by one of two alternative pathways into the mature 5·8S and 25S rRNAs. These pathways are distinguished by the synthesis of either a long or short version of the 5·8S rRNA (the long version is indicated by the black box). The 27S precursors of both pathways are eventually processed at sites C1 and C2 to yield the 7S pre-rRNA and the mature 25S rRNA. Subsequent 3′→5′ exonucleolytic digestion of the 7S rRNA generates the mature 5·8S rRNA. Figure modified with permission from Hong et al. (1997) . Terence I. Moy et al
rRNA genes in most of the eukaryotes are organized as tandem repeats with non transcribed spacers. But individual rRNA genes do have internal transcribed spacers, which are removed during maturation of the pre-rRNA transcript. The diagram shows the individual steps producing specific fragments of certain sizes represented as ‘s’ value.
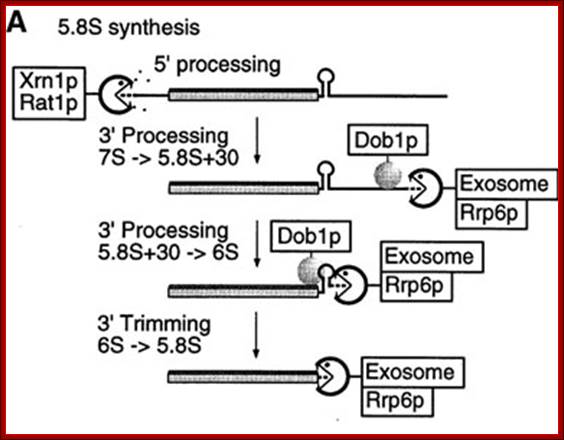
The released 5.8s RNA is processed from 5’ and 3’ ends to generate final 5.8s RNA. The enzymes Xrn1p and Rat1p act as 5’ exosomal enzyme and trim the 5’ end. Similarly the 3’ end is also trimmed by exosome Rrp6p and Dobp (helicase) binds to stem loop structures and removes it and facilitate the 3’ exosome to operate.

rRNA synthesis and processing: http://www.nobelprize.org/
5.8s rRNA Precursor Processing:
- It certain stages of development, ex. During oogenesis in Xenopus, rRNA genes get amplified to thousands of copies by rolling circle mode of replication.
- The amount of rRNA synthesized in a cell accounts for more than 90% of the total RNA found in a cell, which suggests the importance of ribosomal RNA and riboproteins and ribosomes to the cell.
- In most of the eukaryotic systems rRNA primary transcript is substantially larger and contains segments for 18s, 5.8s and 28s RNA blocks and they are in the same order.
- Transcripts don’t contain any tRNAs in the precursors as found in prokaryotic pre-rRNAs.
- The precursor fist cleaved by endonucleases to release small and larger fragments..
- Then the 5’ end of the larger fragment is processed by 5’exonuclease, then cleaved to release the 28srRNA and precursor 5.8srRNA. .
- The 5.8s rRNA is further processed by 3’ exonuceases to generate functional rRNA.
- Sometimes the 3’ extended 5.8s rRNA is incorporated into ribosomes, then it is processed?
Inter cistronic spacer is used for cleaving by endonucleases then the exonucleases trim the 5’ and 3’ ends to generate 5.8srRNA
- The 5s RNA segments are coded for by separate genes located elsewhere, but not in the same region as in nucleolar DNA.
- The 5s RNA genes are transcribed separately, processed and transported into the nucleolus, where the 5s RNA associates with larger rRNA segments during ribosomal assembly.
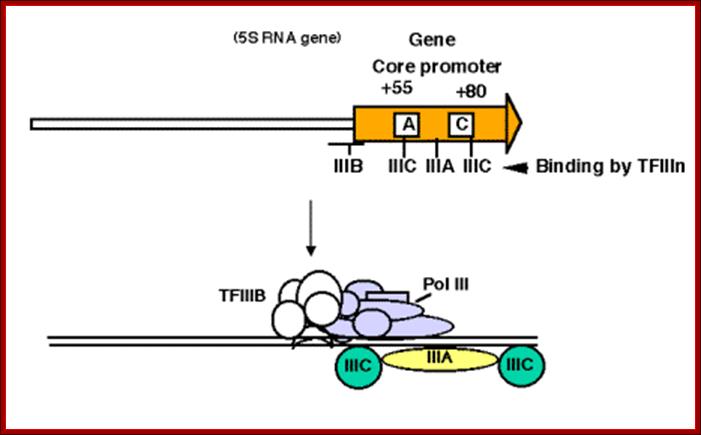
- The 5s rRNA gene promoters are different from others and they are found within the coding region and they use specific type of RNA polymerase i.e. RNA-Pol-III for their transcription, while nuclear rRNA is transcribed by RNA-pol-I.
- 5.8S rRNA, suggesting that this RNA plays a role in ribosome translocation.
5SrRNA:
Unlike prokaryote rRNAs eukaryotes contain specific 5s rRNA genes spread over different chromosomes. The gene structure and transcription is described in the earlier topic.
The mature size of 5srRNA is 120ntds but the precursor is more than 180 ntds. In Xenopus laevis and Arabidopsis there are two categories of 5srRNAs. One is cytoplasmic and the other is nuclear restricted to oocytes or egg development. The cytoplasmic 5srRNA is prevalent during somatic tissue development and in somatic cells. Though the structure of 5srRNA has been established its real function is yet to be elucidated.
In human cells, the cytoplasmic 5S rRNA is imported efficiently into mitochondria, where its role is unclear, but the estimated number of 5S rRNA molecules corresponds roughly with the number of mitochondrial ribosomes.
The precise role of 5srRNA in ribosome function is not fully understood. Based on the results of cross-linking experiments, it was suggested that it may serve as a signal transducer between the Peptidyl transferase center and domain II responsible for translocation, or as a determinant of large-subunit stability. Its importance for the protein biosynthesis machinery was demonstrated in Escherichia coli, in which deletion of more than one 5S rRNA gene greatly impairs the growth rate.
Secondary Structure of 5S rRNA
The 5S rRNA molecule is approx. 120 nucleotides long, with a molecular mass of approx. 40kDa. Regardless of its origin, 5S rRNA can be folded into a common secondary structure. It consists of five helices (I–V), two hairpin loops (C and E), two internal loops (B and D) and a hinge region (A), organized in a three-helix junction. Its general features have been confirmed in a number of structural studies and comparative sequence analyses. In Eukaryote and Archaea, the structure of 5S rRNA is better preserved than in Eubacteria, where much more nucleotide sequence variability is observed.
While the molecule stabilizes the structure of the large ribosomal subunit, 5S rRNA does not contribute directly to any of the active sites in the subunit. 5S rRNA occurs in the ribosomes of the mitochondria of plants and in the ribosomes of their chloroplasts. However, the ribosomes of the mitochondria of fungi and animals lack 5S rRNAs. In humans, the 5S rRNA locus is near the telomere of the short arm of chromosome 1. In Drosophila melanogaster, it is on 2R at 56 E-F. See Chronology, 1963, Rosset and Monier; 1970, Wimber and Steffensen; 1973, Ford and Southern; 1985, Miller, McLachlan, and Klug
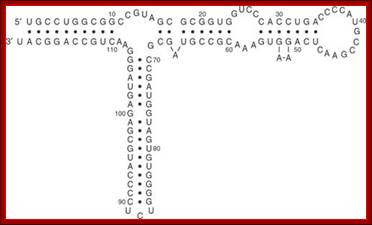
In all organisms, the structure consists of five double-stranded regions (I–V) and five loops (A–E). Loop E, which differs between eukaryotic/archaeal and eubacterial 5S rRNAs, is highlighted in yellow. The palindromic motifs within eubacterial loop E are boxed. Read more: http://www.jrank.org/health/pages/20333/5S-rRNA.html#ixzz1AyQSM513
Copies of the gene for 5S ribosomal RNA. (Birnstiel, Sells, and Purdom, 1972), (Brown and Weber, 1968; Brown, Wensink, and Jordan, 1971). (Ford and Southern, 1973; Wegnez et al., 1972), (Brown and Sugimoto, 1973),
-----I->-----5s------I--------spacer-----I->----5s-----I—spacer-----
Binding of proteins for promoter for RNA polymearase III
The number of copies of this gene in the haploid complement of X. laevis has been reported as 9 000 and 24000. Approximately 1/7 of each sequence codes for the 5S RNA molecule while the remainder of the sequence is considered to be "spacer". Although the many repeating sequences of 5S DNA in X. laevis are similar, they are not identical and thus 5S DNA is a family of related genes. The 5S RNA from several types of somatic cells has been found to be homogeneous in sequence but ovarian tissue synthesizes addition M classes of 5S RNA differing slightly in primary sequence. Moreover the spacer sequences of 5S DNA show some heterogeneity as demonstrated by renaturation studies of the DNA. Based on reconstruction experiments, it was estimated that the 400-bp repeating unit occurs in approximately 1000 copies per haploid genome. Physical characterization of the entire locus was then performed by means of pulsed-field gel electrophoresis.
There are two types of 5s rRNA genes in Xenopus, one Oocyte type and the other is somatic type. The Oocyte-type 5S RNA genes are found at the distal ends of the long arms of most Xenopus chromosomes, including chromosome 9.
- In Xenopus 5s RNA genes are clustered as hundreds of tandem repeats near telomeric region of 18 different chromosomes.
- The gene size is 375 ntds long and processed into 120 ntds long as the final product.
- The 5srRNA genes are transcribed by RNA polymerase III. The precursor is processed to 120 ntd long functional RNA.
- As the nucleolar 45S rRNA transcript is produced, the primary strand goes through certain changes, such as methylation, pseudouridination and formation of secondary structures at specific motifs and cleavages at specified sites.
- These segments with modifications assume stem-loop structures, which are then recognized by RNase III and cut in sequence specific manner. The nucleolus is rich in snU3 RNAs (it has a methylated cap) and RNPs. These Sn U3 RNAs with associated snRNPs are involved in cleavage of pre-rRNAs at defined positions.
- The fragments produced are little longer than the final products, so they are called precursors, p-18s, p-5.8s and p-28s RNAs, which are further trimmed by another set of enzymes.
- The 5.8 s segment gets hybridized at 5’end of the 28s RNA.
- It is during these processing’s riboproteins start associating in stepwise, sequence specific and progressive manner to form ribosomes.
- Processed rRNAs and their associated proteins, in the form of ribosomes, are very stable; their half-life can be any where between 100 days to 120 days or more.
Some rRNA Genes Contain Introns:
- Tetrahymena thermophila is a protozoan ciliate.
- The ribosomal RNA in them is synthesized as 35s long precursor consisting 17s, 5.8s and 26s RNA segments, which are separated by spacers.
- The 17s and 5.8s RNA segments are processed as in other systems, but 26s RNA segment consists of a 413 ntds long extra segment or intervening sequences called introns; until this segment is removed; the RNA fails to organize into ribosome.
- The Intron segment of the 26S RNA is capable of self-cleaving, that means it has the property of Ribozyme.
Precursor rRNA:
Exon Exon
IIIIIIIIIIIIIIIIIII<_____intron_____>IIIIIIIIIIIIIIIIIIIIIIIIIIII__
Intron
Intron structure:
-----U <I 5’p A----------------UpA----------G 3’ I> p U-------
- Distribution of group I introns:
- Mitochondria and plastid genomes of plants and protists (rRNA, tRNA and mRNA genes)
- Nucleus of certain protists, fungi and lichens (rRNA genes)
- Eubacteria (tRNA genes) & phages
- Metazoans - only in mitochondrial genes of a few anthozoans (e.g., sea anemone)
- Structure:
- Some conserved seqeunce blocks
- Processing
- Self-splicing. G attacks 2'OH of RNA
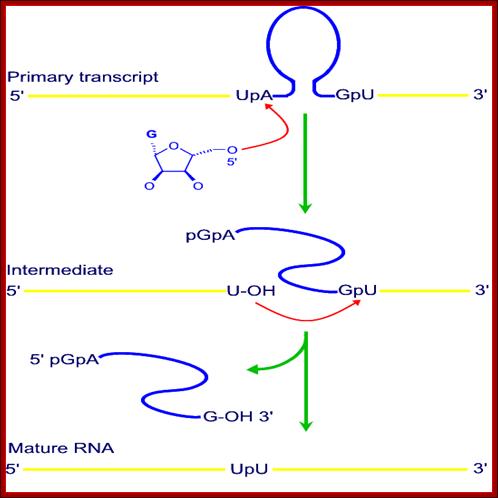
http://nitro.biosci.arizona.edu
· The Exon and Intron splice junction contains a consensus sequence. It also contains a sequence called branching sequence UPA.
· The 5’ joint sequence is 5’U<A. The 3’ joint sequence is G > pU3’. At one third from the 3’ end Intron has a branch sequence 5’ UpA2’OH.
· There are few other sequences in the Intron, which are complementary to the left side of Exon and also nine other internal complementary positions, which pair to each other. Some of the sequences, though not all, act as internal guide sequences (IGS).
· It is because of complementary sequences which drive base paring; the Intron segment undergoes secondary structural, possibly the tertiary structural conformations, which bring the joint sequence very close to each other.
· The folding of the Intron sequence also produces a site for the binding of GTP, GDP or GMP to it as a cofactor.
· Guanine moiety provides negatively charged free 3’OH group to initiate cleaving reaction.
· The Guanines’ OH group, where the O^- carries high negative charge, attacks the phosphodiester bond between U and A, and cleaves the bond between them. In this course of events G itself covalently binds to the 5’end of the Adenine nucleotide of the Intron. Here cleavage and trans-esterification processes are simultaneous.
· This leads to further conformational changes in the intronic RNA, in such a way the free 3’OH group of the left Exon is brought closer to the 3’ Intron-Exon joint, which facilitates cleaving the between G and U and a covalent bond is made with U to generate joined Exon segments (--------U p U------).
· The freed 5’OH group of G, of the Intron reacts with A of the branch point and covalently joins at 2’OH group rather than at 3’.
· Thus the Intron is released as a lariat, which soon gets cleaved into a linear piece and one circular piece with a GpA joint.
- If there is any mutation in the consensus sequences and in IGS, Intron remains unspliced.
- The above-described introns are deemed as group I introns. Such introns are not just restricted to ciliate, such or similar types of introns are also found in mitochondria and chloroplasts, ex. Group-I introns are found in yeast rRNA precursor, but the RNA requires few proteins for splicing, where as in the above said case splicing is performed by the RNA itself, only other requirement is Guanine components.
Ribozymes
- Group I Intron of Tetrahymena thermophila rRNA Genes
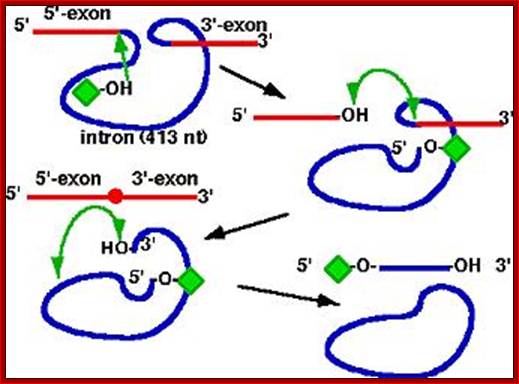
1.pre-rRNA catalyzes its
own splicing
2.2.Phosphodiester transfer reactions
3.first example of a catalytic RNA; termed Ribozyme
4.many other self-splicing group I introns now discovered; http://biocadmin.otago.ac.nz/
The ribosomal intron is 413-416 ntds long. Certain sequences at 5’ end are complementary to 3’ region of the exon. Besides forming base pairing to the 3’end of the exon ( as guide sequence), it also assumes secondary structure so as to produce a site for the binding of G molecule. It is the G molecule that initiates cutting through its negatively charged 2’OH group.
Exon Exon
Precursor-rRNA: IIIIIIIIIIIIIIIIIII<_____Intron_____>IIIIIIIIIIIIIIIIIIIIIII
Intron structure
: ------U <I 5’p A---------------------G 3’ I> p U-------
Spliced Exons:
5’ -----------------------------U p U------------------------- 3’
Released Intron: 5’pA----------------------------------------G3’
- There are other types of Introns, which have their own mechanisms of splicing reactions.
There is a widespread occurrence of spliceosomal introns (69 introns at 27 different sites) in the small- and large-subunit nuclear-encoded rDNA of lichen-forming and free-living members of the Ascomycota.
Borders of such introns are AG-Intron-G. An intron is found within the 16S ribosomal RNA gene of the Archaeon Pyrobaculum aerophilum. An intron is also found in the 23S ribosomal RNA gene of the archaebacterium Desulfurococcus mobilis.
Intron 4 alpha (aI4 alpha) of the yeast mitochondrial COXI gene is a mobile group I intron that contains a reading frame encoding both the homing endonuclease I-SceII and a latent Maturase capable of splicing both aI4 alpha and the fourth intron of the cytochrome b (COB) gene (bI4).
Group I introns work in bacteria too: Experimental evidences for autocatalytic splicing activity have been demonstrated in bacteria, wherein an RNA segment having group-I Intron sequences were introduced in the middle of beta Galactosidase gene by recombinant methods. The constructs are used for bacterial transformation. In the transformed bacterial cells, introduced introns by auto catalytic splicing reactions, the insert is cleaved out and beta Galactosidase segments rejoin to generate a functional beta Galactosidase enzyme, which proves the introduced introns RNA segment is capable of carrying out splicing reaction by itself.