Prokaryotic_Regulation_of_Protein_Synthesis_at_mRNA Level;
Regulation_of_Prokaryotic_mRNA;
RNA degradation:
While many bacteria and mitochondria have polyadenylate polymerases, they also have another type of polyadenylation, performed by polynucleotide phosphorylase. This enzyme is found in bacteria, mitochondria, plastids and as a constituent of the archaeal exosome (in those archaea that have an exosome). It can synthesize a 3' extension where the vast majority of the bases are adenines. As in bacteria, polyadenylation by polynucleotide phosphorylase promotes degradation of the RNA in plastids and likely so in archaea. Polyadenylation of mRNA and noncoding RNAs can lead to destabilization. The poly-(A) tails are short of ~40 nucleotides long on average. In many bacteria, both mRNAs and non-coding RNAs can be polyadenylated. This poly(A) tail promotes degradation by the degradosome, which contains two RNA-degrading enzymes: polynucleotide phosphorylase and RNase E. Polynucleotide phosphorylase binds to the 3' end of RNAs and the 3' extension provided by the poly(A) tail allows it to bind to the RNAs but secondary structure found at 3’ tail would otherwise block the 3' end. Successive rounds of polyadenylation and degradation of the 3' end by polynucleotide phosphorylase allows the degradosome to overcome these secondary structures. The poly(A) tail can also recruit RNases that cut the RNA in two. These bacterial poly(A) tails are about 30-40 nucleotides long.

In Bacillus subtilis, 5′ end-dependent RNA degradation is triggered by RppH; RppH converts the 5′-terminal triphosphate of RNA to a monophosphate ;The resulting intermediate is degraded by the 5′ exonuclease activity of RNase J; This pathway explains the protective effect of 5′-terminal stem-loops in B. subtilis. An RNA Pyrophosphohydrolase Triggers 5′-Exonucleolytic Degradation of mRNA in Bacillus subtilis; Jamie Richards et al; http://www.cell.com/
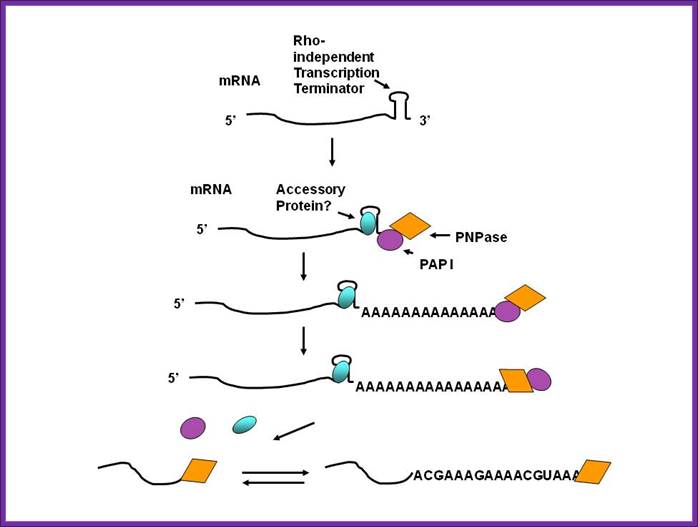
Polyadenylation by Polynucleotide phosphorylase lead to degradation of RNA; http://www.genetics.uga.edu/
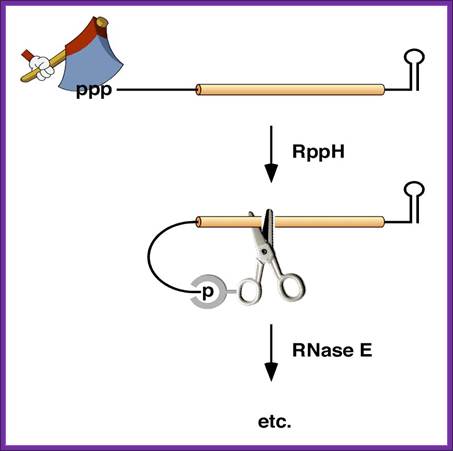
5’ end dependence: In bacteria, the lifetimes of mRNAs can differ by more than an order of magnitude, with profound consequences for gene expression. For many years it had been assumed that mRNA degradation in E. coli begins with endonucleolytic cleavage at internal sites. However, recent findings have challenged that view by showing that mRNA decay can instead be triggered by a prior non-nucleolytic event that marks transcripts for rapid turnover: the rate-determining conversion of the 5' terminus from a triphosphate to a monophosphate. This modification creates better substrates for the endonuclease RNase E, whose cleavage activity is greatly enhanced when the RNA 5' end is monophosphorylated. We have identified the pyrophosphate-removing hydrolase responsible for that 5'-terminal event, the first such bacterial enzyme ever characterized. That the action of the pyrophosphohydrolase is impeded when the 5' end is structurally sequestered by a stem-loop helps to explain the stabilizing influence of 5'-terminal base pairing on mRNA lifetimes in vivo. Interestingly, this master regulator of 5'-end-dependent mRNA degradation in E. coli not only catalyzes a process functionally reminiscent of eukaryotic mRNA decapping but also bears an evolutionary relationship to the eukaryotic decapping enzyme Dcp2; NYU School of medicine; saturun.med.nyu.edu.
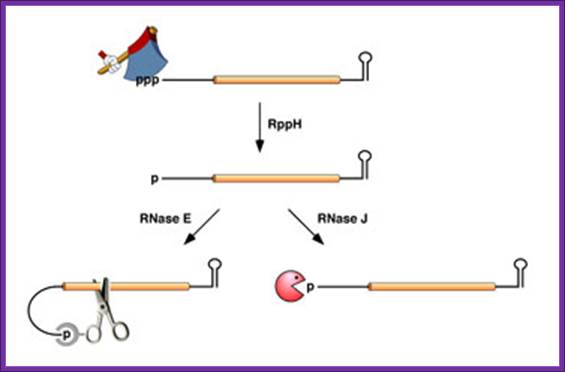
Life time of bacterial mRNA varies by several folds. Joel G. Belasco; http://microbiology-parasitology.med.nyu.edu/
. 
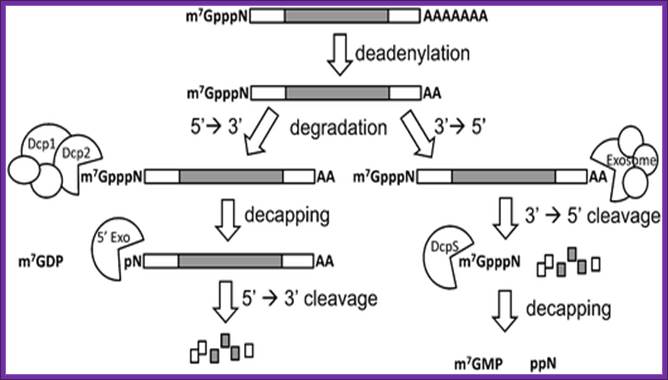
a RNA decay in Escherichia coli. In this pathway, serial internal cleavage by RNase E generates RNA fragments, many of which lack base pairing at the 3′ end. As a result, these degradation intermediates are susceptible to attack by the 3′ exonucleases polynucleotide phosphorylase (PNPase), RNase II, RNase R and (for very short RNA fragments) oligoribonuclease. By contrast, the intact transcript resists exonucleolytic degradation because it is protected by a 3′-terminal stem-loop, which hinders such attack. b RNA decay in eukaryotic cells. In this pathway, poly(A) tail removal by a deadenylase (CCR4–NOT, PAN2–PAN3 or poly(A)-specific RNase (PARN)) yields a deadenylated intermediate susceptible to both decapping by mRNA-decapping enzyme subunit 2 (DCP2) and 3′ exonucleolytic degradation by exosomes. The decapped RNA generated by DCP2 is then degraded by 5′–3′ exoribonuclease 1 (XRN1), whereas the 5′-terminal RNA fragment that results from extensive exosome digestion undergoes cap removal by an alternative decapping enzyme (DCPS) that is specific for oligonucleotides147. These pathways were deduced from early studies of mRNA degradation in E. coli, Saccharomyces cerevisiae and mammalian cells. Ribosomes, PABP and translation factors have been omitted from this figure for simplicity. Joel G. Belasco; Conventional pathways for mRNA degradation in E. coli and eukaryotic cells. Joel G. Belasco; http://www.nature.com/
![Figure 4. Schematic representation of miRNA-mediated mRNA degradation. The AGO2 protein interacts with GW182, whereas the enzymes DCP1/2 remove the 5'cap. Exonucleases start the deadenylation process, leading to the degradation of the mRNA. (Adapted from [11]).](Protein_Synthesis9-Regulation_of_Protein_Synthesis_at_mRNA_Level_files/image006.jpg)
MicroRNA mediated degradation; Schematic representation of miRNA-mediated mRNA degradation. The AGO2 protein interacts with GW182, whereas the enzymes DCP1/2 remove the 5'cap. Exonucleases start the deadenylation process, leading to the degradation of the mRNA.
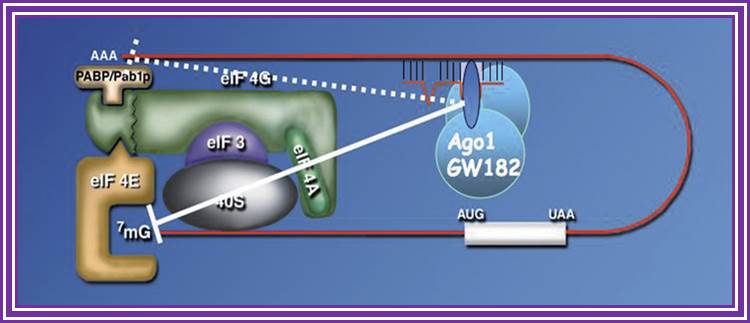
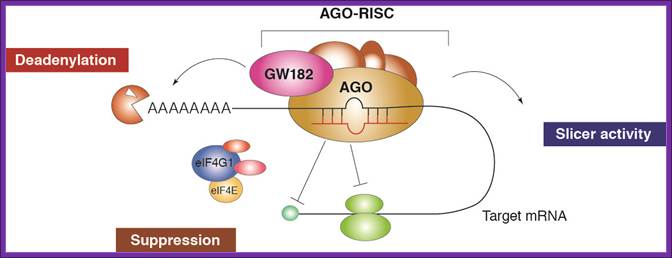
It is generally acknowledged that a single miRNA can deregulate the expression levels of many target genes by a post-transcriptional mechanism [14]. The miRNA-dependent gene expression regulation is mediated by the RNA-induced silencing complex (RISC) containing Dicer and many other associated proteins [15]. RISC incorporates only one functional miRNA strand, and it is often called “miRISC”. Members of the Argonaute (Ago) protein family are central to the RISC function. miRNAs form stable complexes with Argonaute proteins (e.g., AGO2), which represent the core of the silencing complex. However, the binding of miRNAs to mRNA targets can either repress or activate the translation process. The initiation of translation can be inhibited owing to the fact that AGO2 protein binds both the m 7 G cap and the 3'UTR of target mRNA at the same time, therefore preventing the recognition of the cap by eIF4E and the access to mRNA by the translation apparatus (Figure 2). However, many authors observed that miRNAs can affect the translation process not only by inhibiting the translation initiation process, but also intervening at post-initiation level [7,8]. At this level, an additional mechanism of ribosome drop-off is also possible [16]. This process consists in a premature termination of translation and degradation of the incomplete forming protein (Figure 3). The miRNA-mediated mRNA degradation is the result of mRNA terminal deadenylation by 3' → 5' exoribonucleases, 5'-terminal cap removal by decapping enzymes DCP1/2 and hydrolysis of the remaining portion of mRNA by 5' → 3' exonucleases [17]. The DCP1/2 enzymes require the interaction with the RISC components AGO2 and GW182, whereas the interaction between PABPs and GW182 enhances the mRNA deadenylation process, although PABPs are not directly responsible for it (Figure). https://www.researchgate.net
![]()
![]()
RNA Control of Gene Expression ; One of the strategies they have developed to do so is to regulate gene expression at the post-transcriptional level, using RNA and proteins as regulators. Our research interests are (1) to gain insights into the mechanisms by which gene expression in E. coli is modulated, directly or indirectly, by molecules as diverse as small regulatory RNAs (sRNAs) and proteins involved in RNA metabolism such as ribonucleases, poly(A)polymerase, Hfq and possibly others, and (2) to dissect the links between different regulatory networks involving sRNAs and other transcriptional or translational regulators.;www.ibpc.fr
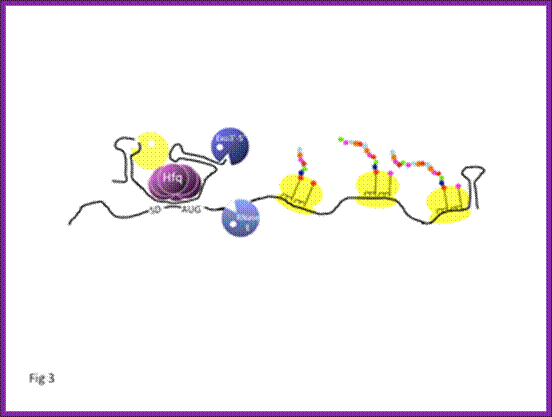
RNA Control of Gene Expression; Bacteria have to respond and adapt very rapidly to environmental stress or changes in growth conditions. One of the strategies they have developed to do so is to regulate gene expression at the post-transcriptional level, using RNA and proteins as regulators. Our research interests are (1) to gain insights into the mechanisms by which gene expression in E. coli is modulated, directly or indirectly, by molecules as diverse as small regulatory RNAs (sRNAs) and proteins involved in RNA metabolism such as ribonucleases, poly(A)polymerase, Hfq and possibly others, and (2) to dissect the links between different regulatory networks involving sRNAs and other transcriptional or translational regulators. Elain Hajnsdorfwww.ibpc.fr
In E. coli, RNA stability depends on endoribonucleolytic cleavages which initiate the process of degradation but also on the addition of poly(A) tails which facilitate the elimination of RNA fragments by exoribonucleases. Other actors, such as RNA helicases, ribosomes, Hfq protein and regulatory s-RNAs also participate in these processes. Our main objective is to understand how these various factors participate in RNA decay; Philippe Régnier and Eliane Hajnsdorf.
![]()

Wilusz² Lab; http://www.eseb.net/
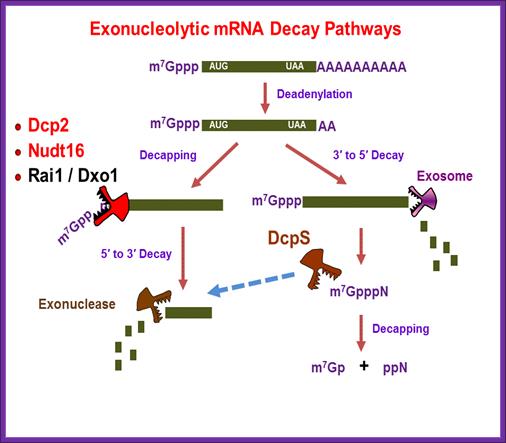
Regulation of mammalian RNA degradation; Kiledjian Lab; Rutger University
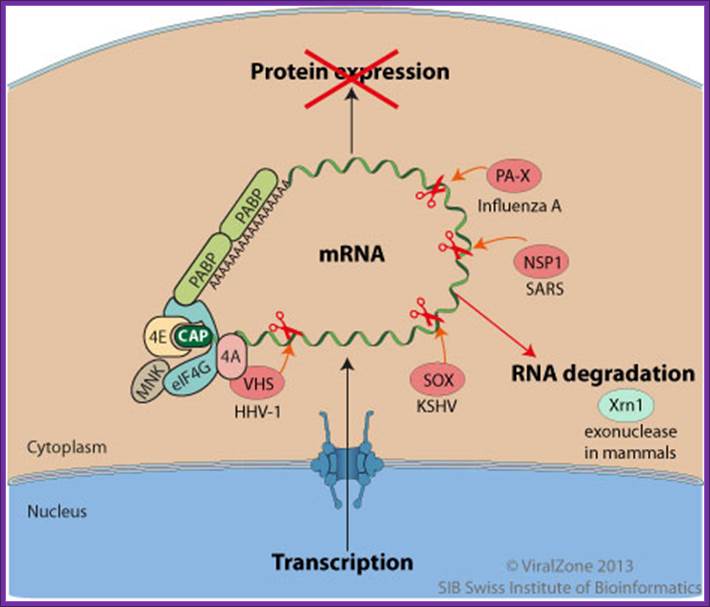
During the lytic replication cycle of Kaposi’s sarcoma-associated herpesvirus (KSHV), SOX proteinpromotes massive cellular mRNAs decay through an endonucleotic internal cleavage of host mRNAs. The resulting RNA fragments are eliminated by the cellular RNA degradation machinery. http://viralzone.expasy.org/

Degradation of bulk mRNA in eukaryotic cells;
Bulk mRNA
decay is initiated by deadenylation (see the figure). There are several
deadenylase complexes in eukaryotes: the PARN2–PARN3 complex is thought to
initiate deadenylation (not shown), which is then continued by the
CAF1–CCR4–NOT complex, the components of which localize to P bodies24. This complex consist of two
deadenylases, CCR4 and CAF1, and the NOT proteins. Following deadenylation,
mRNAs are degraded by exonucleolytic digestion at both ends. Degradation from
the 3' end of the transcript is catalysed by the exosome and the SKI complex25. The 5'![]() 3' mRNA-decay pathway requires
the removal of the cap structure by the decapping enzyme DCP2 and
exonucleolytic degradation by XRN1. Several proteins — DCP1, EDC3, LSm1–7, Pat1
(also known as CG5208) and Dhh1 (also known as RCK/p54, Me31B or CGH-1) — have
been shown to stimulate decapping, but they might function by a different
mechanism. In Saccharomyces
cerevisiae, DCP1 interacts directly with DCP2, whereas in human
cells, a larger complex is thought to exist and includes EDC3 (also known as
LSm16) and Ge-1 (also known as Hedls or RCD-8) (not shown). Most proteins that
are involved in the 5'
3' mRNA-decay pathway requires
the removal of the cap structure by the decapping enzyme DCP2 and
exonucleolytic degradation by XRN1. Several proteins — DCP1, EDC3, LSm1–7, Pat1
(also known as CG5208) and Dhh1 (also known as RCK/p54, Me31B or CGH-1) — have
been shown to stimulate decapping, but they might function by a different
mechanism. In Saccharomyces
cerevisiae, DCP1 interacts directly with DCP2, whereas in human
cells, a larger complex is thought to exist and includes EDC3 (also known as
LSm16) and Ge-1 (also known as Hedls or RCD-8) (not shown). Most proteins that
are involved in the 5'![]() 3' mRNA-decay pathway localize
to P bodies;
3' mRNA-decay pathway localize
to P bodies;
Ana Eulalio, et al;http://www.nature.com/
Synthetic mRNA cap analogs with a modified triphosphate bridge – synthesis, applications and prospects; Jacek Jemielity et al; http://www.rsc.org/

Polycistronic bacterial Mrnas are marked by small black vertical bars; RNaseE is the most abundant RNase, but requires the removal 5 PPP, which is removed by RppH to make the 5’ monoiphosphate. The RNaseP and RNaseH act and finally 5’ and 3’ exonuceases degrade the mRNA; http://www.genetics.uga.edu/
Although the first poly(A) polymerase (PAP) was discovered in Escherichia coli in 1962, the study of polyadenylation in bacteria was largely ignored for the next 30 years. However, with the identification of the structural gene for E. coli PAP I in 1992, it became possible to analyze polyadenylation using both biochemical and genetic approaches. Subsequently, it has been shown that polyadenylation plays a multifunctional role in prokaryotic RNA metabolism. Although the bulk of our current understanding of prokaryotic polyadenylation comes from studies on E. coli, recent limited experiments with Cyanobacteria, organelles, and Archaea have widened our view on the diversity, complexity, and universality of the polyadenylation process. For example, the identification of polynucleotide phosphorylase (PNPase), a reversible phosphorolytic enzyme that is highly conserved in bacteria, as an additional PAP in E. coli caught everyone by surprise. In fact, PNPase has now been shown to be the source of post‐transcriptional RNA modifications in a wide range of cells of prokaryotic origin including those that lack a eubacterial PAP homolog. Accordingly, the past few years have witnessed increased interest in the mechanism and role of post‐transcriptional modifications in all species of prokaryotic origin. However, the fact that many of the poly(A) tails are very short and unstable as well as the presence of polynucleotide tails has posed significant technical challenges to the scientific community trying to unravel the mystery of polyadenylation in prokaryotes. This review discusses the current state of knowledge regarding polyadenylation and its functions in bacteria, organelles, and Archaea. Copyright © 2010 John Wiley & Sons, Ltd.
Regulation of mRNA in Eukaryotes:
Regulation of mRNA in response to iron:
- Iron ions are very important inorganic element involved in a variety of reactions. In most of the cases, it is either bound to protein or forms a structural component of heme. It has a major role in oxygen and carbon dioxide exchange; it is a part of ferrodoxin involved in many enzymatic reactions.
- It is also a part of respiratory system, involved in oxidative phosphorylation and also in photo phosphorylation reactions, thus iron ions have very important cellular functions, so it is required and it is actually required in relatively large amount in comparison to essential micronutrients.
- Excess of iron at extra cellular level is not toxic, but if iron ion concentration, at intra-cellular level, increases, they generate iron free radicals. Such radicals have devastating effects.
- Cells have devised mechanisms to mop up such excess ions and transfer and store excess ions inside the cell.
- Transferrin is an extra cellular protein that is responsible for mopping up iron ions and transfer to Transferrin receptor found on cellular membranes.
- Transferrin receptor is responsible for the transport of iron ions into the cell.
- Ferritin stores Fe2+ ions intracellulary.
Ferritin:
Inside the cell, Ferritin protein is responsible for storing iron ions and release them as and when required. When the concentration of iron ions increases more Ferritin is produced, and when concentration of iron ions decreases, Ferritin concentration also decreases.
- Synthesis of Ferritin protein is controlled at the level of mRNA’s ability to be translated or not be translated.
- Ferritin synthesis is regulated. So also aminoleuvulinate synthase, which performs the first step in heme synthesis, is also regulated.
- The mRNAs of both Ferritin and aminoleuvulinate genes have IRE (Iron Response Elements) loops in their 5’ leader sequence and they don’t have AUUUA sequences, which are the targets for RNA degradation.
![]()
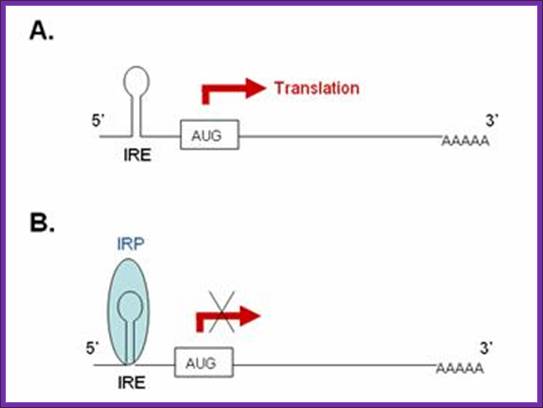
Aconitase; http://www.ebi.ac.uk/
A). In the absence of IRP these transcripts are freely translated. B. Bibding of IRP to the 5’ IRE of the transcript blocks its translation. In iron-deficient cells, the binding of an IRP to an IRE sequence found in the 5’UTR of an mRNA prevents its translation by blocking the mRNA from binding to the ribosome. In iron-replete cells, the absence of IRP binding allows these transcripts to be freely translated. Transcripts that carry IRE in their 5’-UTR include ferritin H and L subunits and aminolevulinic acid synthetase. http://www.ebi.ac.uk/
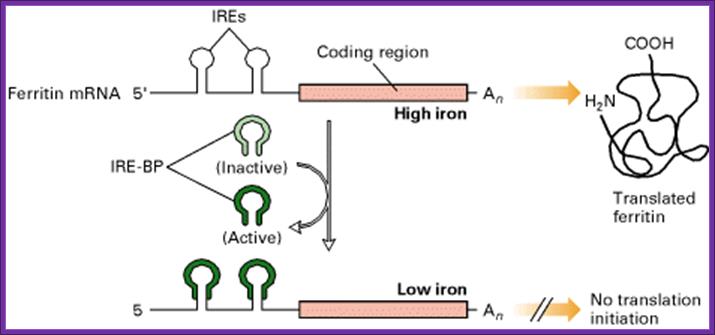
Iron regulatory pathway; http://bioinformatica.upf.edu/
- When the concentration of iron is low in the cell, the IRE-BP (Iron Response Element Binding Protein) binds to the IRE loop, because the loop structure has sequences, which facilitates the binding of IRE-BP, which is nothing but an Aconitase enzyme.
- At lower concentrations of iron, the enzyme protein is in active conformation, so it recognizes the loop structure and binds. The binding of the IRE-Bp protein prevents translation, for it acts as a strong block for the movement of ribosomes along the mRNA. This is what is required, when the concentration of iron is low and there is no need for the synthesis of Ferritin protein, even if it is synthesized it is a waste.
- On the contrary, if the concentration of intracellular iron increases, to mop up and store, it requires more of Ferritin molecules. This is achieved, because when the concentration of iron increases, iron binds to IRE-BP, for IRE-BP has a site for iron. When iron binds to the IRE-BP, it induces conformational change, which renders the protein inactive and falls off from the loop. This allows the translation to proceed beyond this point and Ferritin proteins are synthesized.
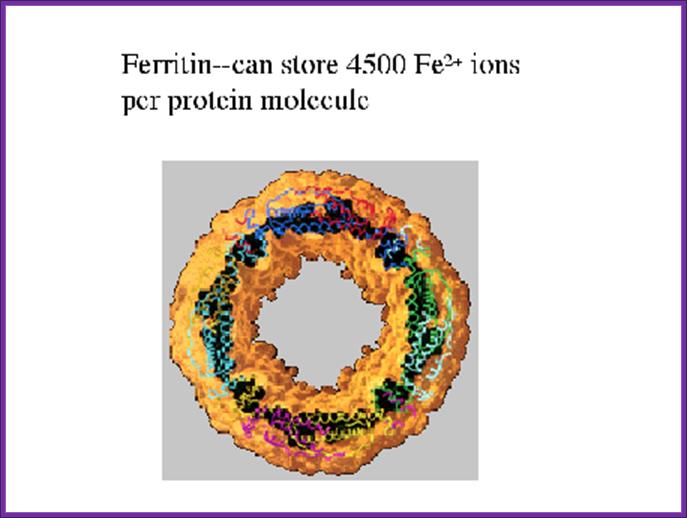
The diagram shows Ferritin that holds all free iron atoms

www.slideshare.net
Transferrin Receptor:
A similar situation exists, when the concentration of intracellular iron is low, it requires the transport of iron ions into the cell.
- Transport of iron into the cell is performed by cell surface iron transferrin receptor.
- When the concentration of iron is low, the mRNA for Transferrin receptor is stabilized and its degradation by digestive enzymes is prevented.
- Transferrin mRNAs, at 3’ noncoding region, contain sequences such as AAUUUUUAA (ARE) sequences in the form of stem loops; there can be more than one such stem loops. Such stem loop structures are sites for RNase. And mRNA degradation is initiated provided they are not protected.
- Degradation of mRNAs makes cells fail to produce any more Transferrin protein, but in a situation where the concentration of iron is low, a protein called repressor IREBP, binds to the stem-loops and prevents RNase to act, thus facilitates the stability of mRNAs and the same are translated to produce more and more of Transferrin receptor proteins, which in turn facilitates import of iron.

On the contrary, if the concentration of iron is more than required, the iron binds to the repressor protein and induces conformational changes and the protein dissociate, making the loop open for digestive action of RNase and the Transferrin receptor mRNA gets degraded and no receptor proteins are produced. This type of regulation acts at the level of stability of mRNA.
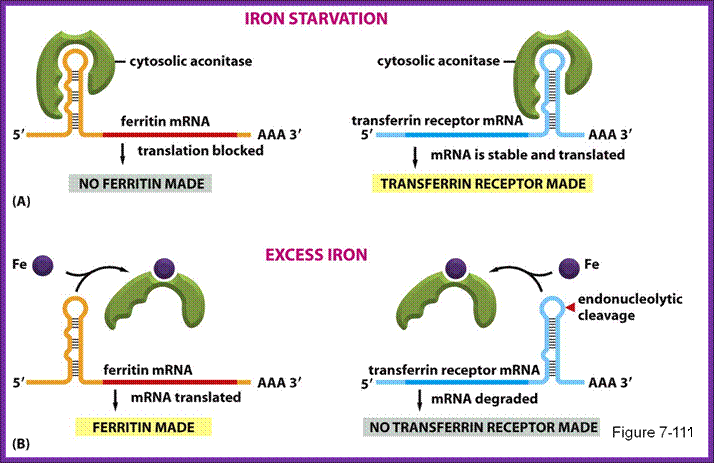
IREBP binds to both Ferritn and Tranferrin IRE elemnets when the concentration of iron very low. When bound yo Ferritin it prevents translation; but when bound to transferring it prevents the degradation of mRNA; .http://www.studyblue.com
Globin protein synthesis and response to Iron:
- In RBC cells iron factor plays an important role in gene regulation either at translation level or at maintaining the stability of their mRNAs. Iron ions are important requirement, for many of the cellular proteins require iron for their activities, ex. Cytochrome and Cytochrome oxidase, hemoglobin, ferrodoxin, Ferritin, Transferrin and many other enzymes.
IRE containing genes and their turnover; (IRE = iron response element); by Mayka Sanchez, PhD;Heidelberg , Germany.
Iron is an essential nutrient
required by almost every organism. Its capacity to exchange electrons makes it
essential for fundamental cell functions, like transport of oxygen and the
respiratory electron transport. However, it is also a potential catalyst for
chemical reactions involving free-radical formation and subsequent cell damage.
Therefore, cellular iron levels have to be carefully regulated inside the
cells.
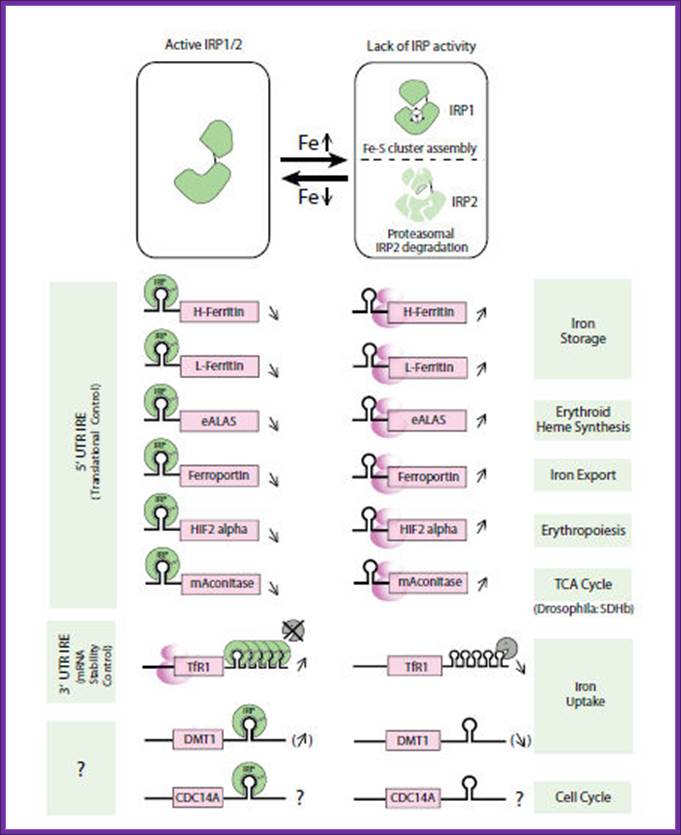
Intracellular iron homeostasis is mainly regulated post-transcriptionally by
the interaction of iron-regulatory proteins (IRP-1 and IRP-2) with non-coding
sequences termed iron-responsive elements (IREs)
present in iron metabolism-related genes, such as transferrin receptor, L- and
H-ferritins, e.g.-ALAS, ferroprotein and divalent cationic transporter 1 (Fig.
1).
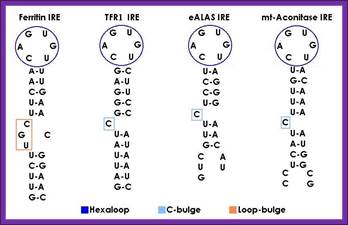
IRE response structural elements found in mRNA coding for iron metabolism proteins: http://bioinformatica.upf.edu/
Iron Responsive Elements Prediction Server by the IMPPC; ;http://ccbg.imppc.org/
What are IREs?
Iron is an essential nutrient required by almost every organism. Its capacity to exchange electrons makes it essential for fundamental cell functions, like DNA synthesis, transport of oxygen and the respiratory chain. However, it is also a potential catalyst for chemical reactions involving free-radical formation and subsequent cell damage and death. Therefore, cellular iron levels have to be carefully regulated inside the cells.
Intracellular iron homeostasis is mainly regulated post-transcriptionally by the IRE/IRP regulatory system. The iron regulatory proteins (IRP1 and IRP2) can recognize a cis-regulatory mRNA motif termed iron responsive element (IRE), a conserved RNA element located in the untranslated regions (UTR) of mRNAs that encode proteins involved in iron metabolism.
A canonical iron responsive element structure is composed of a 6-nucleotide apical loop (5’-CAGWGH-3’; whereby W stands for A or U and H for A, C or U) on a stem of five paired nucleotides, a small asymmetrical bulge with an unpaired cytosine on the 5’strand of the stem, and an additional lower stem of variable length.
To fully understand the complex interplay between players of iron metabolism additional proteins may exist and due to the important role of the IRE/IREBP regulatory network, it is likely that some of these undiscovered genes could contain an IRE .
Strategy to identify new IRE containing genes:
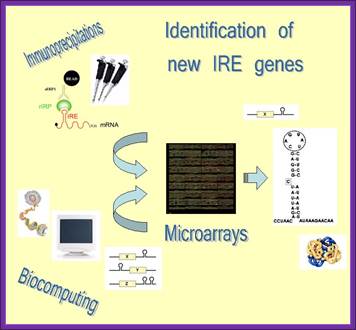
If you want to find out more about iron metabolism and its regulation go to “Dunja or Bruno”.
To identify novel IRE response element containing genes scientists developed an innovative strategy that combines immunoprecipitation, microarray technology and biocomputational approaches. Authors isolated IRE/IRP mRNA-protein complexes by immunoprecipitation experiments and subsequently identified these mRNAs by genome-wide microarray analysis. In a complementary approach IRE-containing mRNAs were selected by biocomputational methods from Genome databases, spotted on the ‘Iron Chip’ cDNA microarray platform and further investigated by hybridization with mRNA isolated from the immunoprecipitation reactions. Using this strategy they have successfully identified novel IRE containing genes and currently they are working on the regulatory mechanisms that the IRBPs play on these candidates via their IRE sequence “Dunja or Bruno”.
Regulation Histone mRNA;
Histone mRNAdoes not possess any polyA tail. But processed in different ways. Histone mRNA regulation controlled to its cell cycle events. Replication of the eukaryotic chromosome requires synthesis of not only DNA, but also histone proteins needed to package the newly replicated DNA into chromatin. During transition from G1 to S phase transcription of histone genes increase by fivefold, for the histones are required for the assembly DNA into chromatin threads.
Metazoan
histone mRNAs are unique: their pre-mRNAs contain no introns, and the mRNAs are
not polyadenylated, ending instead in a conserved stem-loop structure. In
Drosophila, canonical poly(A) signals are located downstream of the normal
cleavage site of each histone gene and are utilized when histone 3' end
formation is inhibited. Here we define a subcomplex of poly(A) factors that are
required for histone pre-mRNA processing. We demonstrate that Symplekin,
CPSF73, and CPSF100 are present in a stable complex and interact with
histone-specific processing factors. We use chromatin immunoprecipitation to
show that Symplekin and CPSF73, but not CstF50, cotranscriptionally associate
with histone genes. Depletion of SLBP recruits CstF50 to histone genes. Knockdown
of CPSF160 or CstF64 downregulates Symplekin but does not affect histone
pre-mRNA processing or association of Symplekin with the histone locus. These
results suggest that a common core cleavage factor is required for
processing of histone and polyadenylated pre-mRNAs.
A core complex of CPSF73,
CPSF100, and Symplekin may form two different cleavage factors for processing
of poly(A) and histone mRNAs. (Jun 10, 2017). https://www.researchgate.net/

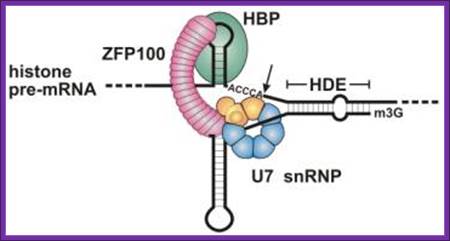
The sequence and predicted configuration of the mouse H2a-614 pre-mRNA (top) and the mouse U7 snRNA (bottom) during 3′ end processing. The base pair interaction between the two RNAs is indicated with vertical lines (Watson–Crick base pairs) and dots (GU base pairs). The tri-methyl guanosine cap (TMG) at the 5′ end of the U7 snRNA is framed. The unique U7 Sm site, the histone downstream element (HDE) and the site of cleavage are indicated. The region of the U7 Sm site that UV cross-links with Lsm11 is labeled with the asterisk. Zbigniew Dominski,William F. Marzluff.
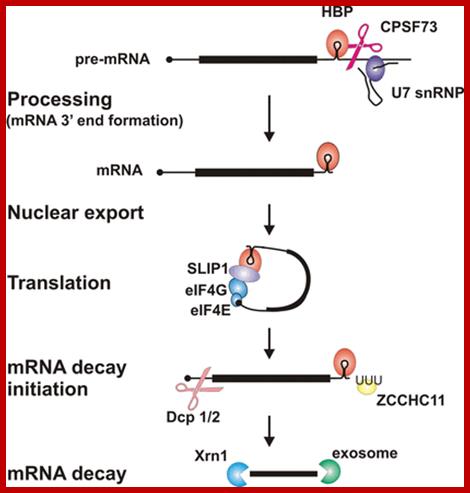
Key steps and factors in the post-transcriptional control of histone gene expression
Histone mRNA 3′-end processing requires the RNA-binding protein HBP, which binds to the conserved hairpin structure in histone pre-mRNA, and the U7snRNP, which binds to a sequence element downstream of the cleavage site. Together with other factors they position the nuclease CPSF73 for cleavage to produce histone mRNA ending immediately after the hairpin. After nuclear export, HBP interacts with SLIP1 and other translation initiation factors to form a closed-loop structure for efficient translation. This structure is disrupted, presumably when histone mRNA decay is initiated, for example at the end of S-phase. Addition of an oligo(U) tail by the terminal uridylyl transferase ZCCHC11 is an early step in decay, which involves decapping followed by 5′→3′ degradation by Xrn1 or 3′→5′ decay by the exosome. http://www.biochemsoctrans.org
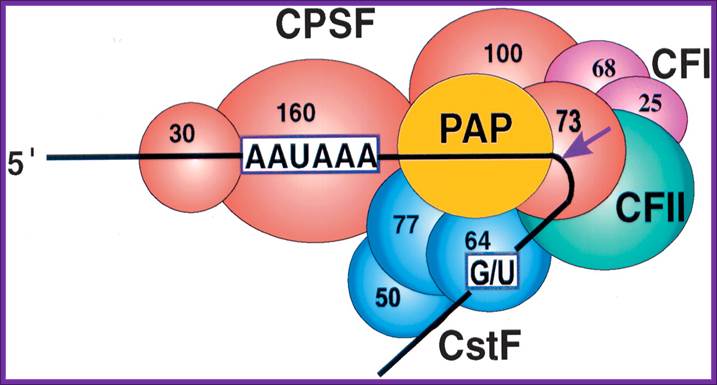 CPSF-73 as the 3′ processing nuclease. The core
polyadenylation machinery (e.g., RNAP II is not included) is illustrated as it
would assemble in a precleavage complex on an mRNA precursor, with CPSF-73 at
the site of cleavage. The AAAUAA and GU-rich signal sequences are indicated,
and the arrow denotes the cleavage site. The protein–protein and protein–RNA
interactions depicted are consistent with available data. See text for details
of protein factors. http://rnajournal.cshlp.org
CPSF-73 as the 3′ processing nuclease. The core
polyadenylation machinery (e.g., RNAP II is not included) is illustrated as it
would assemble in a precleavage complex on an mRNA precursor, with CPSF-73 at
the site of cleavage. The AAAUAA and GU-rich signal sequences are indicated,
and the arrow denotes the cleavage site. The protein–protein and protein–RNA
interactions depicted are consistent with available data. See text for details
of protein factors. http://rnajournal.cshlp.org

CPSF-73 as the 3′ processing nuclease. The core polyadenylation machinery (e.g., RNAP II is not included) is illustrated as it would assemble in a precleavage complex on an mRNA precursor, with CPSF-73 at the site of cleavage. The AAAUAA and GU-rich signal sequences are indicated, and the arrow denotes the cleavage site. The protein–protein and protein–RNA interactions depicted are consistent with available data. See text for details of protein factors;CPSF 73 is mRNA 3’ processing endonuclease http://rnajournal.cshlp.org/
Cleavage and Polyadenylation Specificity Factor (CPSF):
A protein with 160 kDa and 30 kDa (zinc finger) RNA binding subunits, which binds to the AAUAAA signal upstream of the cleavage site. Additional 73 kDa and 100 kDa subunits do not contact RNA and their function is unknown; http://www.hixonparvo.info/

Known components of the 3′ end processing machinery cleaving histone pre-mRNAs. The position of symplekin in the complex is hypothetical and the presence of CPSF-100 has not been supported experimentally. U7sn RNA is basepaired to the 3’ end of the histne mRNA. Both have stem loop sructures. Histone stem loop structure are bound by SLBP proteins. SLBP2 is Oocyte specific and SLBP1 is cytoplasm specific. As oocytes mature SLBP2 gets degraded. XSLBP2 is an inhibitor of translation while SBP1 activates translation.Note it is discerned that stored histone mRNA has a short oligo-A tail which are removed as oocytes mature. And xSLBP2 is degraded (x=Xenopus). mRNA with oligo A-tails are not translated, but their translation is stimulated in the presence of xSLBP1, Symplekin is Histone 3’ end processing protein. Ricardo sanchez and W.F Maraluff.;Marcela Dávila López and Tore Samuelsson have identified both metazoan and protozoans contain short poly-A tail required for 3’ processing.
Mutational analysis of the region of Lsm11 required for the interaction with FLASH. (A) Sequences and factors involved in 3 Ј -end processing of histone pre-mRNA. The HDE that base pairs with the 5 Ј end of the U7 snRNA (thick gray line) is underlined, and the site of cleavage located 5 nucleotides after the stem-loop is indicated with an arrow. The fragment of histone pre-mRNA located downstream of the cleavage site is referred to as the DCP. (B) Sequence alignment of the N-terminal half of human (top) and Drosophila (bottom) Lsm11. Residues of human Lsm11 that were changed to alanines are underlined. The PL dipetide located in positions 33 and 34 is referred to as PL1, whereas the more-downstream dipeptide at positions 155 and 156 is referred to as PL2. (C and D) Pulldown of 35 S-labeled wild-type and mutant versions of the N-terminal fragment of Lsm11 by GST or GST fused to the N-terminal fragment (amino acids 1 to 139) of human FLASH, as indicated. https://www.researchgate.net

Representation of the processing reaction occurring at the 3′ end of replication-dependent histone precursor mRNAs. The endonucleolytic cleavage (indicated by the scissors) occurs between the highly conserved hairpin (see also Fig. 3A) and the purine-rich sequence HDE. Trans-acting factors are hairpin binding protein (HBP), the U7 snRNP and zinc finger protein 100 (ZFP of 100 kDa). Sophie Jaegera, Sharief Barendsb et al.
Molecular mechanism of histone pre-mRNA 3’ end processing by U7 snRNP; U7 snRNPs contain two unique Lsm proteins, Lsm10 and Lsm11 (replace D1 and D2), which are required for histone RNA processing. Lsm1-7 form a cytoplasmic rather than a nuclear complex that mediates RNA decapping and degradation. Lsm2-7 associates with snR5 snoRNA required for the site-specific pseudouridylation of rRNA. https://www.ebi.ac.uk
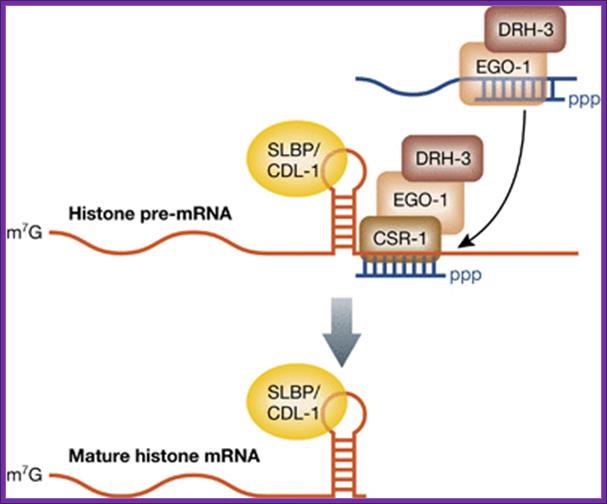
Birthing of histone mRNA by CSR1 section; The 3′‐end processing of histone mRNAs by the CSR‐1 RNAi pathway. The RdRP EGO‐1, along with DRH‐3, synthesizes endo‐siRNAs, which load onto the Argonaute CSR‐1. Endo‐siRNAs that pair perfectly to sequences near the stem loop in histone pre‐mRNAs recruit CSR‐1, which facilitates cleavage and maturation of the mRNAs. The SLBP, called CDL‐1 in C. elegans, functions in 3′‐end formation and remains associated with the histone mRNA. Amy E Pasquinelli; http://emboj.embopress.org/
Histone genes encode for pre‐mRNAs that undergo maturation through a mechanism distinct from all other RNA Pol II transcripts by substituting polyadenylation with an alternative 3′‐end formation pathway (Marzluff et al, 2008). The 3′‐end of a histone pre‐mRNA is marked by a short stem‐loop followed by a histone downstream element (HDE). The hairpin is bound by the stem loop binding protein (SLBP) and the HDE is recognized by the U7 small nuclear RNA (U7 snRNA) through base pairing interactions. Next, cleavage and polyadenylation specificity factor 73 (CPSF73) joins the complex and cleaves the pre‐mRNA a few nucleotides downstream of the stem loop. SLBP remains associated with the processed histone mRNA and fulfills roles normally attributed to the polyA binding protein, such as promoting nuclear export and translation. Additionally, worms lack an HDE‐like sequence after the stem loop in histone mRNA. One solution to this problem is presented in a new study, where RNAi‐related factors regulate the formation of histone mRNA 3′‐ends (Avgousti et al, 2012). Instead of utilizing an HDE to position the cleavage complex, sequences downstream of the stem loop in histone pre‐mRNAs were found to be complementary to endogenous small interfering RNAs (endo‐siRNAs). These endo‐siRNAs are produced by the RNA‐dependent RNA polymerase (RdRP) EGO‐1 and associate with the Argonaute CSR‐1; Amy E Pasquinelli.
Mechanisms
of maternal mRNA regulation: implications for mammalian early embryonic
development
Anilkumar Bettegowda1, 2, George W. Smith1, 2, 3 Michigan State University, East Lansing, MI
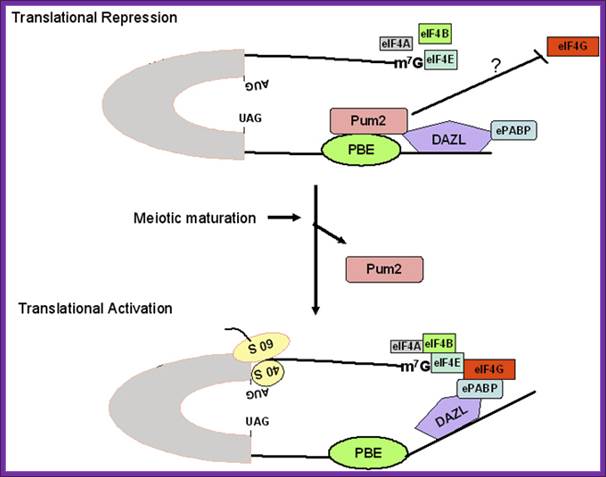
Figure 1. Schematic model for regulation of pumilio-2 (Pum-2) bound RINGO/Spy mRNA translation during oocyte maturation. The RINGO/Spy mRNA is repressed by the binding of Pum-2 to the pumilio-binding element (PBE) in the 3' untranslated region (UTR). Pum2 resides in a complex consisting of embryonic poly (A)-binding protein (ePABP) and the RNA binding protein deleted in Azoospermia-like (DAZL) and interferes with the interaction between translation initiation factors (e.g., eIF4G) and the 5' end of the mRNA. During meiotic maturation, Pum-2 dissociates from the protein complex bound to mRNA, and thereby ePABP interacts with eukaryotic initiation factor-4G (eIF4G) at the 5'end, resulting in translational activation. http://www.bioscience.org/

Figure 2. Schematic model for regulation of cytoplasmic polyadenylation element (CPE)-containing maternal mRNA during oocyte maturation. CPE-binding protein (CPEB), an RNA binding factor binds to CPE sequence and further interacts with Maskin, a repressor protein that prevents cap-dependent translation. Maskin acts as a eukaryotic initiation factor-4E (eIF4E) binding protein and competitively binds to eIF4E at the same region as eukaryotic initiation factor-4G (eIF4G) and thereby prevents the access of translation initiation factors to the 5' end of the mRNA. CPEB further interacts with three other proteins; (a) Cleavage and polyadenylation specificity factor (CPSF), a protein bound to poly(A) signal; (b) symplekin, a scaffold protein; and (c) germline development deficient-2 (GLD2), a cytoplasmic poly(A) polymerase. During meiotic maturation, RINGO/Spy protein acts as a cofactor for cyclin dependent kinase (CDK) activity and presumably activates Aurora-A kinase, which in turn activates CPEB by phosphorylation. CPEB phosphorylation leads to activation of CPSF/Symplekin/GLD2 protein complex, which in turn elongates the short poly (A) tail of CPE-containing transcripts. Maskin undergoes differential phosphorylation at many residues by CDK and thereby releases the repressive effects of maskin at the 5'end of the mRNA. The longer poly(A) tail binds to embryonic poly(A) binding protein (ePABP) and in turn recruits eIF4G to the cap-structure of the mRNA consequently activating translation; http://www.bioscience.org/
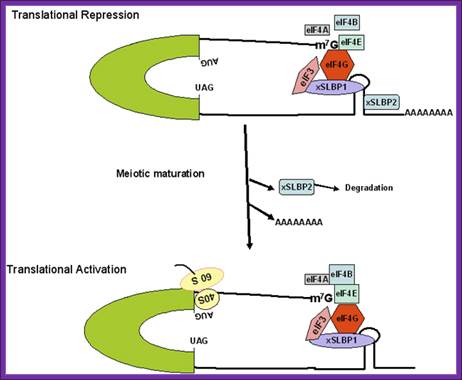
Figure: Schematic model for regulation of histone mRNA during oocyte maturation: In immature Xenopus oocyte, histone mRNAs have a short oligo(A) tail attached to the conserved stem-loop structure. At 5’ end it is bound by e4E and e4G. The stem-loop sequence is bound to two stem loop binding proteins (xSLBP1 and xSLBP2). The oligo (A) tail and the binding of xSLBP2 to the stem-loop sequence and 5’ e4G inhibit translation initiation. During meiotic maturation, the oligo (A) tail is removed, the xSLBP2 is degraded by unknown mechanisms and histone mRNA is translated. Symplekin and multiple other polyadenylation factors participate in 3′-end maturation of histone mRNAs.; http://www.bioscience.org/
Drosophila UNR regulation:

Model for the role of Drosophila UNR in the regulation of msl-2 mRNA translation: Model for the role of Drosophila UNR in the regulation of msl-2 mRNA translation. The female-specific protein SXL binds to poly(U) stretches in the 5′ and 3′ UTRs of msl-2 mRNA. SXL bound to the 3′ UTR recruits UNR to adjacent purine-rich sequences. The SXL/UNR complex inhibits the association of the 43S ribosomal complex with the 5′ end of the mRNA, thereby repressing translation. SXL bound to the 5′ UTR inhibits the scanning of ribosomal complexes that may have escaped the UNR block (Beckmann et al. 2005). In male flies, which lack SXL, UNR does not bind to the 3′ UTR of msl-2 mRNA and translation proceeds. http://genesdev.cshlp.org
The female-specific protein SXL binds to poly-(U) stretches in the 5′ and 3′ UTRs of msl-2 mRNA. SXL bound to the 3′ UTR recruits UNR to adjacent purine-rich sequences. The SXL/UNR complex inhibits the association of the 43S ribosomal complex with the 5′ end of the mRNA, thereby repressing translation. SXL bound to the 5′ UTR inhibits the scanning of ribosomal complexes that may have escaped the UNR block. In male flies, which lack SXL, UNR does not bind to the 3’UTR msl2 mRNA and translation proceeds, (Beckmann et al. 2005).
Stored mRNAs:
In many situations the mRNA that is produced is processed and transferred into cytoplasm; for example developing oocyte. In the oocyte they are transported to specific sites within the cell, either at the anterior or posterior or antero-posterior location with the help of motor proteins and cytoskeleton elements. Example in the case of Drosophila oocyte, before fertilization Bicoid, Hb, Nanos, Gurken and few others are maternal messengers and they are transported into the developing oocyte and positioned in specific loci. That is how the positional information is determined in oocytes. This is not unusual, for this mRNA localization and activation of mRNA is prevalent in all cells. The said mRNA are not translated but remain inactive because the poly (A) is shortened and bound by specific factors and make the mRNA as an inactive but reserve informosomes. After fertilization, poly-(A) is added and the mRNA protein complexes become active. This type of inactive storage mRNAs are also found in developing or developed seeds in plants. In fact seeds store a large number of such preprogrammed mRNAs for they require when the dry seeds absorb water and wakeup for germination.
Inactivation and activation of mRNA:
In oocytes of Xenopus, Drosophila and other systems such as dry seeds mRNAs are stored for the future use; till then they remain in inactivated state by shortening of Poly-A tail to 25-50ntds. In addition these mRNA have certain sequence elements such as Cytoplasmic Polyadenylation Elements CPE to which CPEB proteins bind. Binding of Maskin brings eF4E and CPEB together, this prevents translation. Upon fertilization, signals activate translation by phoshorylation of CPEB, which gets freed from the complex. This leads to the recruitment of CPSF, and Poly-A polymerase, which adds A nucleotides to original length; thus protein synthesis is renewed.

Schematic model for regulation of cytoplasmic polyadenylation element (CPE)-containing maternal mRNA during oocyte maturation. CPE-binding protein (CPEB), an RNA binding factor binds to CPE sequence and further interacts with Maskin, a repressor protein that prevents cap-dependent translation. Maskin acts as a eukaryotic initiation factor-4E (eIF4E) binding protein and competitively binds to eIF4E at the same region as eukaryotic initiation factor-4G (eIF4G) and thereby prevents the access of translation initiation factors to the 5' end of the mRNA. www.ekspresihati.info; www.sfsoo.com
Oocyte stored truncated mRNAs:
During oocyte development, a large population of maternally-encoded mRNAs is synthesized and stored for “use” in particular stages of development. These maternal mRNAs typically possess short poly (A) tails (20-40 nts) and are not available for translation (being “masked” by a complex of RNA-binding proteins). During oocyte maturation or following fertilization, these masked mRNAs are de-masked and get polyadenylated. Thus they get activated for translation. This activation is a regulated process that helps to coordinate the ballet of gene expression attendant with meiotic maturation and early development. As such, it touches on many tangential phenomena (such as movement of stored mRNAs). Symplekin is heat labile component containing CPSF ad other factors required for cytoplasmic poly-A adenylation and translation activation in Xenopus oocyte maturation.

Model of MK2 regulation of TTP-directed and CCR4-CAF1 catalysed deadenylation of ARE-containing mRNA. In its unphosphorylated state, TTP binds an AU-rich element (ARE) found in inflammatory mRNAs and promotes shortening of the poly-(A) tail by recruiting the CCR4-CAF1 deadenylase complex. The CCR4-CAF1 complex comprises the large central core protein CNOT1, which binds seven other subunits together with the deadenylase subunits, CAF1 and CCR4. It is not known which subunit directly interacts with TTP. Activation of the p38 MAPK pathway leads to phosphorylation of Ser-52 and Ser-178 of TTP by MK2, which decreases the interaction of TTP with the CCR4-CAF1 complex and inhibits deadenylation thereby increasing inflammatory gene expression (Marchese et al., 2010); Jonathan Dean, Daniel Bulbrook and Hannah Brazier. http://www.kennedy.ox.ac.uk/
Stored mRNA in Arabidopsis thaliana: Nakabayashi K, Okamoto M, Koshiba T, Kamiya Y, Nambara E. Beltrán-Peña E, Ortíz-López A, Sánchez de Jiménez E. Sano N, Permana H et al. Mdelseny,etal. Dry seeds of A. thaliana contain ~12000 different species of mRNA, most of them expressed from genes with ABRE response elements. Embryonic seeds contain large number of stored ribosomal protein genes. In rice dry seeds contain Actinomycin D resistance potential for germination but not to cycloheximide. It means dry seeds store mRNAs required for their germination to some extent. Store seed embryos contain Poly-A mRNA which gradually disappear during water imbibition, but soon mRNA appear due to new transcription and they contained poly-A tails in Radish. The length of poly-A tail appears to be short.

Kidney bean sprouting (Biology Encyclopedia).
EJC in nonsense mediated mRNA-decay:
During the second step of splicing in eukaryotic cells, the EJC is deposited approximately 20-24 nucleotides from the 5’ end upstream of the splice junction (where two exons are joined), when the lariat has formed and the exons are ligated together. EJC components are RNPS1, Y14, SRm160, Aly/REF and Magoh, RNPS1 can function as a coactivator of splicing, but along with Y14, it also takes part in the process of nonsense-mediated decay in eukaryotes. SRm160 is another coactivator that has been proposed to enhance mRNA 3’ end processing. The protein component Magoh is thought to facilitate the sub cytoplasmic localization of mRNAs while Aly is engaged in nuclear mRNA export. Aly is believed to be recruited to the exon junction complex by the protein UAP56 (WikI). UAP56 is recognized as an RNA helicase but acts as a splicing factor required for early spliceosome assembly. Another factor involved in the EJC pathway is DEK. This component is known to take part in a variety of functions ranging from splicing to transcriptional regulation and chromatin structure. The binding of the EJC to the mRNA occurs in a sequence independent manner, to form the mature messenger ribonucleoprotein (mRNP). The EJC remains stably bound to this mRNP as it is exported out of the nucleus and into the cytoplasm. Protein components are either bound to or released by the EJC as it is transported. In order for the translocation of mRNAs through the nuclear pore complex to occur, a heterodimer consisting of NXF1/TAP and NXT1/p15 must be bound to the transcripts. NXF1/TAP is a major receptor for the export of mRNAs to the cytoplasm. This is due to the fact that it interacts with both RNA-binding adapter proteins and components of the nuclear pore complex.
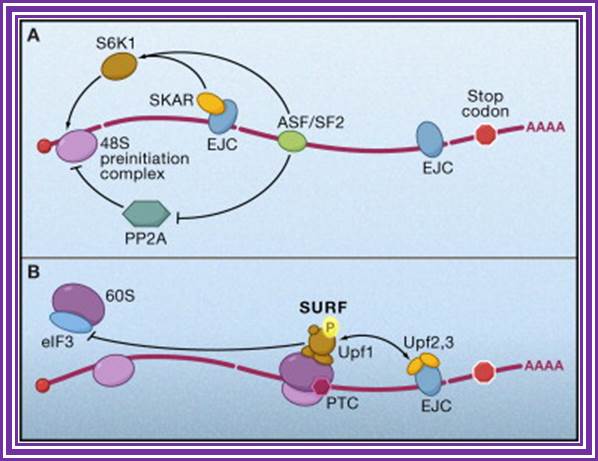
Figure. Splicing Factors Regulate Translation Initiation. Effects of splicing-dependent mRNP components on translation initiation (A) Both the exon junction complex (EJC) and ASF/SF2 bound to spliced mRNA can promote the first or “pioneer” round of translation by recruiting 40S ribosomal protein S6 kinase 1 (S6K1), a component of the TOR signaling cascade. For the EJC pathway, this is accomplished via the EJC-interacting protein SKAR. ASF/SF2 can also promote translation initiation by inhibiting the S6K1 antagonist PP2A phosphatase.(B) When an EJC is located more than 50 nucleotides downstream of a premature termination codon (PTC), interaction between the EJC and SURF complex causes phosphorylation of Upf1, which then inhibits additional rounds of translation by an interaction with the translation initiation factor eIF3.
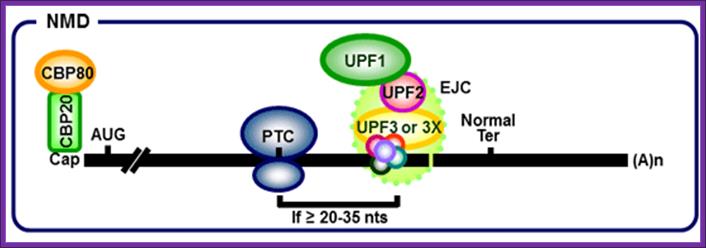 When ribosomes while translation meets the EJC complex, the EJC
complex is released. When the ribosome moves further and encounters a Nonsense
codon as PTC it stops and recruits termination factors. The location of an
exon–exon junction >50 nucleotides downstream then sets the stage for the
interaction between the termination complex and EJC that provokes NMD. In both yeast and human cells, the major pathway for mRNA decay
is initiated by the removal of the 5’ cap followed by
degradation by XNR1, an exo ribonuclease enzyme. The other pathway by which
mRNA is degraded is by deadenylation
from 3’-5'. The NMD pathway in human cells comprises the factors UPF1, UPF2,
UPF3A, UPF3B, SMG1, SMG5, SMG6 and SMG7. Presence of nonsense codon in the
middle leads to the assembly of a complex composed of UPF1, SMG1 and
the release factors, eRF1 and eRF2, on the mRNA. UPF1 comes into contact with
UPF2 and UPF3, triggering the phosphorylation of UPF1.
The phosphorylated UPF1 then interacts with SMG-5, SMG-6 and SMG-7, which
promote the dephosphorylation of UPF1. SMG-7 is thought to be the terminating
effector in NMD, as it accumulates in P-bodies, which are
cytoplasmic sites for mRNA decay. In both yeast and human cells, the major
pathway for mRNA decay is initiated by the removal of the 5’ cap followed by
degradation by XNR1, an exo-ribonuclease enzyme. The other pathway by which
mRNA is degraded is by deadenylation
from 3’-5'. Dr. Robert Singer: http://www.urmc.rochester.edu/
When ribosomes while translation meets the EJC complex, the EJC
complex is released. When the ribosome moves further and encounters a Nonsense
codon as PTC it stops and recruits termination factors. The location of an
exon–exon junction >50 nucleotides downstream then sets the stage for the
interaction between the termination complex and EJC that provokes NMD. In both yeast and human cells, the major pathway for mRNA decay
is initiated by the removal of the 5’ cap followed by
degradation by XNR1, an exo ribonuclease enzyme. The other pathway by which
mRNA is degraded is by deadenylation
from 3’-5'. The NMD pathway in human cells comprises the factors UPF1, UPF2,
UPF3A, UPF3B, SMG1, SMG5, SMG6 and SMG7. Presence of nonsense codon in the
middle leads to the assembly of a complex composed of UPF1, SMG1 and
the release factors, eRF1 and eRF2, on the mRNA. UPF1 comes into contact with
UPF2 and UPF3, triggering the phosphorylation of UPF1.
The phosphorylated UPF1 then interacts with SMG-5, SMG-6 and SMG-7, which
promote the dephosphorylation of UPF1. SMG-7 is thought to be the terminating
effector in NMD, as it accumulates in P-bodies, which are
cytoplasmic sites for mRNA decay. In both yeast and human cells, the major
pathway for mRNA decay is initiated by the removal of the 5’ cap followed by
degradation by XNR1, an exo-ribonuclease enzyme. The other pathway by which
mRNA is degraded is by deadenylation
from 3’-5'. Dr. Robert Singer: http://www.urmc.rochester.edu/
Exon junction complexes play a major role in mRNA surveillance. More specifically, they are found in the nonsense mediated decay pathway (NMD). In normal mRNA translation, the ribosome binds to the transcript and begins amino acid chain elongation. It continues on until it reaches the location of the exon junction complex, which it then displaces. Next, translation is complete when the ribosome reaches a termination codon. In NMD, the mRNA transcript contains a premature termination codon (PTC) due to a nonsense mutation. If this codon occurs prior to the EJC site, then mRNA decay is triggered. The EJC and its position serve as a type of regulator, determining whether the transcript is defective or not.
EJCs are also known to take part in NMD in another way; the recruitment of the surveillance factors UPF1, UPF2 and UPF3. These proteins are the most important components of the NMD mechanism. The EJC protein MAGOH, Y14 and eIF4AIII provide a binding for UPF3, which acts as a bridge between UPF2 and UPF1 forming a trimeric complex. Within this complex, UPF2 and UPF3 act cooperatively to promote ATPase and RNA helicase of UPF1. The EJC core stably anchors the UPF complex to the mRNA, and aids in regulation of essential UPF1 protein. Ribosomes which are stalled on a PTC recruit UPF1 through interactions with the release factor eRF1 and eRF3. Along with the protein SMG1, eRF1, eRF3 and UPF1 form the complex SURF. This complex forms a bridge between the ribosome and the downstream EJC which is associated with UPF3 and UPF2. This interaction triggers the phosphorylation of UPF1 by SMG1, causing the dissociation of eRF1 and eRF3. The complex produced consists of EJC, UPF3, UPF2, phosphorylated UPF1 and SMG1 and in turn triggers degradation of the mRNA.
In mammalian cells, nonsense-mediated mRNA decay (NMD) degrades mRNAs that terminate translation sufficiently upstream of a post-splicing exon junction complex (EJC) of proteins. Components of the EJC include the NMD factor Upf1. We have found that Upf1 also interacts with the RNA binding protein Staufen (Stau)1. Stau1 mediates the decay of specific cellular mRNAs when translation terminates normally. This Stau1-mediated mRNA decay (SMD) contributes significantly to the network of post-transcriptional regulatory pathways in mammalian cells. SMD differs from NMD because it occurs independently of splicing and after down-regulating Upf NMD factors other than Upf1. However, SMD is similar to NMD because it requires translation and the recruitment of Upf1 sufficiently downstream of a termination codon, Madam Curie Bioscience.
Nonsense Mediated decay is involved in detection and decay of mRNA transcripts which contain premature termination codons (PTCs). PTCs can arise in cells through various mechanisms: germline mutations in DNA; somatic mutations in DNA; errors in transcription; or errors in post transcriptional mRNA processing. Failure to recognize and decay these mRNA transcripts can result in the production of truncated proteins which may be harmful to the organism. By causing decay of C-terminally truncated polypeptides, the NMD mechanism can protect cells against deleterious dominant-negative, and gain of function effects. PTCs have been implicated in approximately 30% of all inherited diseases; as such, the NMD pathway plays a vital role in assuring overall survival and fitness of an organism.
A surveillance complex consisting of various proteins (eRF1, eRF3, Upf1, Upf2 and Upf3) is assembled and scans the mRNA for premature stop codons. The assembly of this complex is triggered by premature translation termination. If a premature stop codon is detected then the mRNA transcript is signaled for degradation – the coupling of detection with degradation occurs.
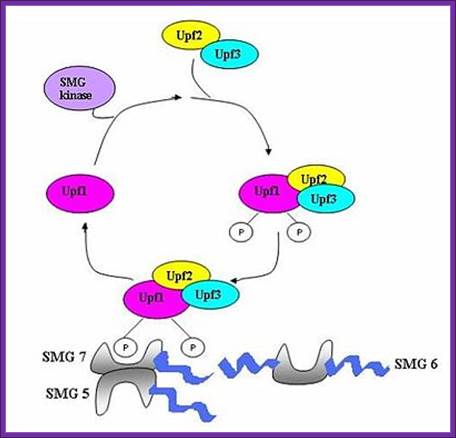
UPF1 is a conserved helicase which is phosphorylated in the process of NMD. This phosphorylation is catalyzed by SMG1 kinase. This process requires UPF2 and UPF3. Dephosphorylation of UPF1 is catalyzed by SMG5, SMG6 and SMG7 proteins; http://en.wikipedia.org/

Nonsense mediated decay in mammals is mediated by the exon-exon junction. This junction is marked by a group of proteins which constitute the exon junction complex (EJC). The EJC recruits UPF1/SMG by transcription factors eRF1/eRF3. Interactions of these proteins lead to the assembly of the surveillance complex. This complex is ultimately responsible for the degradation of the nonsense mRNA. http://en.wikipedia.org/

Nonsense mediated mRNA decay in invertebrates is postulated to be mediated by the presence of a faux 3' untranslated region (UTR). These faux 3'UTRs are distinguished from natural 3'UTRs which follow natural stop codons. This is due to the lack of binding proteins which are normally present in natural 3'UTR. These binding proteins include the poly(A)-binding protein (PAPB).(Weeki); http://en.wikipedia.org/
Nonstop Mediated Decay:
Often mRNA get broken and thus they lack terminal TER codon. This can be due to broken mRNA or premature Poly adenylation. In such cases ribosomes move up to the end or up to poly-A and stops. Thus large number of ribosomes initiated gets stranded on the mRNA.

Nonstop mediated mRNA. Translation of a mRNA without a stop codon results in the translation of the ribosome into the 3' poly-A tail region. this results in a stalled ribosome. The ribosome is rescued by two distinct pathways. The mechanisms are dependent of the absence or presence of the Ski7 protein; http://en.wikipedia.org/
The ribosome is rescued by two distinct pathways. The mechanisms are dependent of the absence or presence of the Ski7 protein. This pathway is active when Ski7 protein is available in the cell. The Ski7 protein is thought to bind to the empty A site of the ribosome. The Ski7 is now associated with the nonstop mRNA and it is this association which targets the nonstop mRNA for recognition by the cytosolic exosome. The Ski7-exosome complex rapidly deadenylates the mRNA molecule which allows the exosome to decay the transcript in a 3’ to 5’ fashion (WIKI).
No-Go Decay:
Because of strong secondary structure in the middle of mRNA or anywhere associated with some proteins blocks the ribosomal function and halts its progress. This also leads to jamming of ribosomes on mRNAs. Dom34/Hbs1 likely binds near the A site of stalled ribosomes and may facilitate recycling of complexes. In some cases, the transcript is also cleaved in an endonucleolytic fashion near the stall site; however the identity of the responsible endonuclease remains contentious. The fragmented mRNA molecules are then fully degraded by the exosome in a 3’ to 5’ fashion and by Xrn1 in a 5’ to 3’ fashion

Nonstop mediated mRNA. Translation of a mRNA without a stop codon results in the translation of the ribosome into the 3' poly-A tail region. this results in a stalled ribosome. The ribosome is rescued by two distinct pathways. The mechanisms are dependent of the absence or presence of the Ski7 protein.. Blocked ribosome on mRNA recruits endonucleases which cut the mRNA after the stalled ribosome. Once RNAs are released they are degraded by different RNases. http://en.wikipedia.org/wiki/MRNA_surveillance
Most of the above mentioned mRNAs finally find their place in P-bodies, where they are degraded by various means of RNases.
mRNA,s regulated by RNA interference:
RNAi is a fortuitous discovery; it has changed the concepts of small noncoding RNA and their importance. Many of them are small interference RNAs. They are 21-23nts long derived from long dsRNA or stem-loop structured RNA precursors.
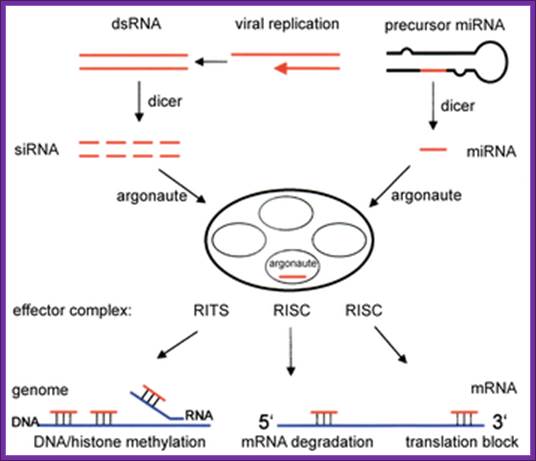
RNA interference is a process of gene silencing that plays an important role in development and maintenance of the genome. The RNAi pathway is complex. It is initiated by an enzyme called ‘Dicer’ which cleaves double stranded RNA (dsRNA) into 20-25 bp fragments. An RNA-induced silencing complex (RISC) is then formed by base pairing between complementary mRNA and 1 of the 2 strands of each new fragment. This formation of the RISC complex is followed by degradation of the complementary mRNA by the endonuclease argonaute. Argonaute is the catalytic component of the complex. The short 20-25 bp fragments are known as small interfering RNA (siRNA) can be artificially introduced and microRNA (miRNA) can be produced endogenously. RNA interference has become a valuable research tool since it allows the prevention of translation of specific genes by introducing siRNA or mi RNA complementary to the mRNA one wishes to suppress. This is as good as gene knock out process.

miRNAs use two mechanisms to exert gene regulation. Some animal miRNAs can bind to mRNA targets with exact complementarity and induce the RNAi pathway. miRNAs also bind to targets with imperfect complementarity and block translation. There is no evidence that C. elegans miRNAs use the former pathway.
Monica C. Vella, Frank J. Slackhttp://www.wormbook.org/
The long dsRNA is cut and processed by Dicer group of enzymes. The short pieces of RNA get associated with RiSC complexes and release one of the strands. Such strands specifically bind to 3’UTR of certain mRNA which have complementary sequences and induce degradation of mRNA, thus silence a gene transcript; there are two modes- one leads to the destruction of bound mRNA another inhibit translation of the mRNA.
Degradation of mRNAs in Response to Viral infection:
RNAi Pathway –miRNAs/siRNA/piRNA;
In 1996 the Nobel Prize was awarded to Professors Andrew Fire and Craig Mello, for their discovery of “small interfering RNAs” (siRNAs). They discovered that some non-coding regions of RNA played critical roles in the regulation of gene expression. Instead of coding for the creation of proteins, these RNAs acted as an off switch, de-activating mRNAs. Specifically, these short RNA sequences formed short double stranded regions (surprise! only DNA was supposed to be double-stranded) that were recognized by a protein complex and cut in half. Then the protein/RNA complex proceeded to do something truly shocking. Instead of dropping the cut (and now presumably useless) RNA strands, the protein complex retained one of the strands, and the two together romped around the cell binding to a specific target mRNA sequence and cutting them up. Far from useless – the non-coding RNA regions turned out to be very powerful regulators of gene expression. As a single mRNA may act as the template for hundreds or even thousands of proteins, a regulatory switch that can destroy all mRNAs from a specific gene can quickly and effectively terminate the effect of that gene within a cell (an activity termed RNA interference, or RNAi).
Small Interfering RNAs: Patent Law’s Response to a Revolutionary Invention; by James Lafave; http://www.ipbrief.net/
RNAi Pathway –miRNAs/siRNA/piRNA; http://en.wikipedia.org/; http://ruo.mbl.co.jp/
RNA interference (RNAi) is a mechanism that controls the gene expressions in a sequence specific manner. Small ncRNAs, including miRNAs, siRNAs as well as piRNAs, function as the guide molecules that incorporated into RNA-induced silencing complex (RISC). They control the translation and degradation of their target mRNAs that have the complementary sequences with their guide strands. RNAi has recently been shown to play an important role in a variety of biological phenomena including the dynamics of early development, morphogenesis, cell growth and tumorigenesis. Small ncRNA does not function directly by itself. It functions only when it is incorporated into Argonaute (AGO) protein, a key component of RISC. It functions as a guide to recognize the target mRNA. The translational inhibition and degradation of the target mRNA are due to the activities of the RISC components including AGO and GW182. Recently, post-transcriptional regulation through these small ncRNAs is attracting the attention of researchers; James Lafave