Genetics of Cancer - III
Mechanism:
Basis for Transformation:
Cells in normal course of replacement of cells or growth divide and redivide. Cell division can lead to more cell divisions but in a programmed way. But during replacement, cells generated by cell division get differentiated into its mother tissue type. Here cell division is controlled and regulated. The number divisions and the number of cells required are controlled.
1 Cell division in a cell cycle progresses through a series of biochemical and molecular events in a well-defined way, which are manifested in physical changes; in terms of cell size, chromosomal morphology, chromosomal DNA replication and duplication of chromosomal threads into sister chromatids, disappearance and reappearance of nuclear membrane, appearance of Mitotic apparatus, binding of tractile fibers to Kinetochore regions of chromosomes, chromosomal movement to central equatorial region and then to polar regions, reappearance of nuclear membrane and finally division of cytoplasm to generate two cells. Every single event which is physically observable has a series of progressive biochemical and molecular steps that eventually manifests in such changes.
2 The most important stage in cell division is Interphase and Mitosis (M-phase); mitosis consists of Prophase, Metaphase, Anaphase, and Telophase where nuclear division ends and cytoplasmic division takes over. In some, one finds only nuclear division e.g. early nuclear division of at early endosperm or in some fungi only nuclear division- coenocytic. Interphase intercalates between two M-phases. In some cases such as early embryogenesis, where cell cycles complete in about 20 to 30 minutes, M-phase directly leads to S-phase without any intervening Interphase.
3 But in normal cell divisions, Interphase occupies nearly ~20hrs in a 24 hr cell cycle. In cells, which are going through cell divisions, Interphase goes through certain phases of biochemical and molecular activities, which are manifested, into chromosomal duplication and they are named as G1, S and G2 phases. In a developed tissue, most of the cells remain in quiescent stage where cells remain in Go where cell remain silent, which is also called a resting stage. This is the stage at which most of the cells go through differentiation and remain in differentiated cell types for a long time. Cells in such state get activated to divide when required for cell replacement or stimulated to divide.
4 Quiescent cells can be induced to enter into cell division mode and the factors responsible are called Mitogens, which work through signaling mechanism. In unicellular cellular system, just sufficient nutrition status provides signals to act at the cell surface level. In both types, the signal transduction takes place in a cascade fashion, leading to a variety of activation and inactivation of cellular components including gene activation. These processes which act in a sequence and temporal but regulated way. These, in turn, propel the cell into cell division mode.
5 During cell cycle events, each of the stages are controlled and regulated by checkpoints, such as G1 to S stage and G2 to M and M stage itself. Checkpoints monitor the early events and only when all processes are completed then allow progress to the next stage, otherwise they are held up till all the events are completed. Each other stages have several events and the progression is regulated. Even M-stage has regulatory events. However once they pass on to M stage, cell division is irreversible. Similarly induction of M-phase is held in check before completion of DNA replication and checks for the completion of repairs of any errors in replication or DNA damage. Some of the genes and gene products involved in these events are briefly cited.
6 If any one of the genes and their products which promote the progression of the cell division overcoming the check points are disabled by mutation (loss of function), or by another mutation (gain of function) can lead to uninhibited cell division leading to a mass of cells or tumours. Deregulation of checkpoint genes or its products render cell cycle out of control and cells multiply uninhibited. Many cellular cell cycle regulating genes, if undergo mutation making them to over express, or constitutively express can lead to proliferation of cells in unlimited fashion. Even cell cycle inhibitors or suppressors, if become incapable of their timely functions, the cell cycle regulation gets deranged and proliferation takes over. Many genes involved in signal transduction pathway, which generate a cascade of events, such as kinase, and kinase-kinase, if they undergo changes, can have a profound effect on cell cycle.
7 It is not just one mutation of this sort is enough for causing cancer; for the cancer to manifest in all its glory, with time, the cell has to suffer many more such genetic abuses. It is a cumulative effect and the aging augments such facility. But modern way of life, with added pollution and food habits can act as carcinogens; with this the incidence of cancer in population is rising.
8 Mutations in the genome can cause a variety of disturbances in the normal function of genes or gene products. Not all of them are able to induce unhindered proliferation of cells. Any one of the following or combination of mutations can cause tumorigenesis.
9 Mainly mutation in genes responsible for regulating cell cycle, genes involved in suppressing cell cycle progression and genes that generate transcription factors, activators, co activators of transcription, are responsible for cancer. Mutations can be of loss of function (negative) or gain of function (positive) or double negative mutants with gain of function. Mutations can be in the form of point mutations, deletions, translocations and duplications; all can lead to loss of function or gain of function.
10 Genes that are responsible for inducing cancer are called Oncogenes. And counter part of Oncogenes are actually normal functional cellular genes, they are called Proto Oncogenes or C-Oncogenes. Some of the cellular genes, carried by viruses, which cause cancer on their infection, are called V-Oncogenes ex. V-Src. Many viruses have genes that can induce cancer on infection are called viral Oncogenes. Three hundred or more Oncogenes have been identified so far. That number can be more. Several of them are trans-membrane proteins and several of them are signal-transducing factors, many of them are transcriptional factors and there are tumorigenic suppressor and activator genes. The list of cancer causing genes never ends as long as animals, especially human beings exist on this planet. Plants are no exception to cancers, they have many but mostly ignored for they don’t affect human lives.
Outline:
Cells in Resting State:
In embryonic state, developing cell population divides and redivide only to determine and differentiate into adult cell types as tissues. Among them some have potential to divide and regenerate, and they are called stem cells. Cells in resting state are mitotically at Go stage.
1 Cell division is controlled and regulated, where once cells divide the daughter cells differentiate and develop into specific cell type and never indulge in continuing cell division. It is genetically programmed and inherent.
2 In adult tissues, cells remain in quiescent state or what is called Go state. They will be induced to divide when replacement is required.
3. In cells at Go stage, all the genes and gene products required for initiating and progressing into cell division mode are disabled or held in check by many mechanisms.
1 Signaling transduction pathway leading to activation of cell cycle events remains silent for there are no signal inputs and no signal transduction to cytoplasmic components.
2 RB and its associated factors sequester transcriptional initiating or activating factors and they are not available for activation of genes required for initiating DNA replication. RB in its unphosphorylated state acts as tumor suppressor. Related proteins such as p107 and p130 also sequester such transcription factors. RB is a tumor suppressor.
3 The gene products such as cyclins for activating Cdk kinase are not made available. Cyclin and Cdks are engines of cell cycle. They are not synthesized. Many such Cdk-cyclins are held in check by a variety of CKIs called Cdk-cyclin inhibitors.
4 Many cellular kinases and phosphatases required for initiating cell division remain silent or inactive.
5 Tumor suppressor gene products such as P53 and P21 hold back cell division when there is damage to DNA, this control is important for if the cell enters into cell division mode when the DNA is damaged; it will end up in abnormality.
6 Many transcriptional factors, co-activators and its associated factors required for cell division remain inactive when cells are in resting stage.
7 Anaphase Promoting Complex (APC) is inactive and the surveillance system is fully operative.
Cell Cycle in Operation:
1 After completion of cell division, the differentiating cells enter into Go state. All the factors, that are active hitherto, are shut off. So the cell develops its cell specific characteristic shape and functions; remarkable indeed!
2 Such cells become active only when the cell division is required, or they can be induced to go through cell division by growth factors or mitogens.
3 Before initiation of cell division, cells have to grow to a size and acquire cell mass that is internally measured by sensors inside the cell; when the cell mass to cell volume ratio is reached a threshold, the cell is ready for cell division. Yet they don’t enter into cell division mode, they wait for signals.
4 Signals can be mitogens or nutritional factors; they act on cell surface receptors, which perform signal transduction through specific protein kinase systems such as Tyrosine or Serine/Threonine protein kinases.
5 Kinases are the key components in cell cycle events. Similarly several phosphatases also play critical functions in time dependent manner.
They act on cellular components, depending upon the target, and the targets may become active or inactive. This can have a cascading effect. i.e. few more kinases become active, where a kinase kinasing another kinase and each have their own targets.
a. Some such cellular kinase phosphorylates a variety of target components such as Cdk-Cyclin inhibitors, RBs and specific Transcriptional factors, all required for cell cycle.
b. Among them certain cyclin-dependent kinase catalytic subunit gets associated with its specific Cylins to become dimers (Cdk-cyclins); first it is made inactive wee1 kinase by phosphorylating Thr 14 (or Tyr 15). Cdk-Cyclin activating kinase CAK phosphorylates Thr 160 near the active site, yet the dimer is inactive because of phosphorylation at Thr14/Tyr15 of CDK kinase subunit. However signal transduced kinases such as Ras-kinase phosphorylates Cdc 25, and activates it. Cdc-25 is a phosphatase. The active Phosphatase removes the Phosphate group from Thr 160 (Thr161). This act makes the Cdk-cyclin active. There are different sets of Cdk-cyclin dimers, which get activated in the same way but at different times in temporal fashion. The timing of activation and inactivation of these factors is the key for cell division to progress or to halt.
3. The Major Cyclins and Cdks of Vertebrates and Budding Yeast;
|
CYCLIN-CDK |
VERTEBRATES |
BUDDING YEAST |
||
|
COMPLEX |
CYCLIN |
CDK PARTNER |
CYCLIN |
CDK PARTNER |
|
G1-Cdk |
cyclin D* |
Cdk4, Cdk6 |
Cln3 |
Cdk1** |
|
G1/S-Cdk |
cyclin E |
Cdk2 |
Cln1, 2 |
Cdk1 |
|
S-Cdk |
cyclin A |
Cdk2 |
Clb5, 6 |
Cdk1 |
|
M-Cdk |
cyclin B |
Cdk1** |
Clb1, 2, 3, 4 |
Cdk1 |
|
Phase |
Cyclin |
CDK |
|
G0 |
C |
Cdk3 |
|
G1 |
D, E |
Cdk4, Cdk2, Cdk6 |
|
S |
A, E |
Cdk2 |
|
G2 |
A |
Cdk2, Cdk1 |
|
M |
B |
Cdk1 |
|
Species |
G1 |
G1/S |
S |
M |
|
Cln3 (Cdk1) |
Cln 1,2 (Cdk1) |
Clb 5,6 (Cdk1) |
Clb 1,2,3,4 (Cdk 1) |
|
|
Puc1? (Cdk1) |
Puc1, Cig1? (Cdk1) |
Cig2, Cig1? (Cdk1) |
Cdc13 (Cdk1) |
|
|
cyclin D (Cdk4) |
cyclin E (Cdk2) |
cyclin E, A (Cdk2,1) |
cyclin A, B, B3 (Cdk1) |
|
|
X. HYPERLINK "https://en.wikipedia.org/wiki/Xenopus_laevis"laevis |
either not known or not present |
cyclin E (Cdk2) |
cyclin E, A (Cdk2,1) |
cyclin A, B, B3 (Cdk1) |
A list of CDKs with their regulator Proteins and others;
• CDK4; cyclin D1, cyclin D2, cyclin D3
• CDK5; CDK5R1, CDK5R2. See also CDKL5.
• CDK6; cyclin D1, cyclin D2, cyclin D3
• CDK9; cyclin T1, cyclin T2a, cyclin T2b, cyclin K
• CDK10
4. At early G1 the RB and their related proteins such as p107 and p130 bind E2F (E2F1 to E2F 5) family of transcription factors and they are held back from activating many genes required for initiating DNA replication. But when RB is phosphorylated, RBs and its associated factors release all the E2F members. They in turn activate transcription of S-phase factors. Many RB associated dimers protein (DPs) are also activated. This is critical for the cell transition from G1-to S phase. Till the required factors such as DNA polymerase, helicases and others are made available cell does not enter into S-phase.
5. At this point of time, if there is any damaged DNA, it is detected by p53 tetramer protein and halts the progress into the next stage; thus it suppresses cell cycle progress.
6. When the required replication factors are made available, replication is initiated. The critical part of replication initiation is firing of replication origin. This is done by ORC (Origin Recognition Complex, a hexamers ~400KD) binding to A1 region of the ORE (Origin Recognition Elements) and provides pre-initiation inputs. At this point acquiring of licensing factors, such as Mcm (Mini Chromosomes Maintenance factor) and Cdc6, Cdt1 and Geminin is very important. Geminin prevents reinitiation of S-phase before the completion of M-phase. Phosphorylation of Cd6 is important for Cdc6-p is responsible for the loading of Mcm as pentamer, one at each of the replication fork. This assembly of preinitiation complex is essential for the buildup of replication machinery. These licensing factors are acquired at the previous stage i.e. when the nuclear membrane disassembles and reassembles at the M-phase. The licensing factors are acquired only once in one cell cycle, without it replication is not initiated. As the s-phase in progress G1-cyclins and S-phase cyclins are degraded by SCF mediated ubiquitination method.
7. Further activation of another set of Cdk-Cylin components takes place when cyclin-B enters the nucleus that activates the cell to leave G2 and enter M-phase. This kinase is often called as maturation promoting factor (MPF) or mitosis promoting kinase (MPK).
8. If there is any replication error or if the replication is not completed or the DNA is damaged, p53 halts the progression into M-phase till the DNA is fully restored. P53 activate p21, which binds to Cdk-Cyclins, and inhibit its activity. If the DNA damage is beyond repair, p53 induces the cell to go through programmed cell death, called Apoptosis.
9. Once the G2-M-phase checkpoint is crossed, there is no return and the cell goes through various steps of M-phase leading to completion of cell division. Activation of this complex is crucial for the entry into M-phase. This activated MAP-Kinase phosphorylates target proteins like lamins of nuclear membranes, H1 histone tails of chromatin, Securins of Cohesin complex, Nucleophosmin (Centrosome), Microtubules and Microfilaments (?) and APC (Anaphase Promoting Complex).
10. Phosphorylation of lamins leads to dismemberment of nuclear membrane and pore complex as membrane vesicles, phosphorylation of H1 leads to condensation of chromosomes into short and stable chromosomes; perhaps proteins such as Condensins have an important role in chromosomal condensation. Phosphorylation of Cytosomes leads to duplication of centrosome and start organization mitotic apparatus. Furthermore microtubules that underlay the network of endoplasmic reticulum undergo dissociation into monomeric Tubulins. Thus the whole ER membrane dissociates into vesicles; even the microfilaments reorganize and reorient in the cellular milieu.
11. The most important effect of MAPK is the activation of APC complex. It is a protein complex of 8 subunits. It is activated by MPK first by an adaptor protein called Cdc20. This activated APC targets Cohesin complex, where the Securin is ubiquitinated and fed into proteasome for degradation. When the Securin is degraded, it frees the sequestered and activated Separin. Separin is a protease (endoprotease), which digests Scc1p, which is an important component of Cohesin that is responsible for gluing chromatin threads together and the kinetochore yet undivided. Degradation of Scc1p releases the chromatin strands free and also releases the kinetochores into free structures for the organization of tractile fibers for anaphase movements. Then the adaptor protein Cdc20 is displaced by another protein called Cdh1, which activates the APC complex to target M phase-cyclin A and later M-cyclin B. Degradation of these cyclins marks the end and exit of M-phase, But it also reverses what MAPkinase induced dissociation of nuclear membrane, condensation chromatin, dismemberment of MTs and others. Reversing the process is the appearance of Nuclear membranes with their respective pore-complexes, chromosomes relaxes, MTs reassemble into Mitotic spindle fibers and tractile fibers attach to kinetochore complex; thus all components and complexes were restored. Then chromosomes are pulled to their respective poles and finally cytoplasm divides, by the activated cytoplasmic components.
12. Thus the cell cycle executes its operation to produce two daughter cells equal amounts of genetic material.
13. The whole process is highly regulated with many check and surveillance systems. Any mistake or malfunction in these events can lead either to the death of the cell or the daughter cells acquire abnormal or unstable genome; thus it can lead to malfunction of the cells, where the cell can undergo cell division in uncontrolled fashion to produce a mass of cells; it is what one calls cancer.
14. In normal cell cycle event the important components that control the cell cycle in an orderly fashion are mitogens, mitogens activated membrane receptors, receptor mediated signal-transducing system, the cytoplasmic transducing system; most of them are kinases and kinase-kinases, and their targets. Another set of components is CDK kinase regulators such as cyclins; wee1 kinases, Cdc 25 phosphatases and the most important component are kinase inhibitors, and tumor suppressors such as RBs and p53. Malfunctioning of any of them or a combination of any of them can lead to repeated cell cycles without any check into abnormal growth of cells, proliferation and cancer.
Events during Tumorigenesis:
1 During repeated cell divisions, in situations like in vitro cultures, Telomeric DNA shortens and perhaps Telomerase activity is also lost. This leads to genetic instability what is called crisis; because of this, cells can go through uncertainty. If such cells reactivate or regain telomerase activity, the cell goes through repeated cell divisions; the said state is called Immortal state. If the damage is severe and beyond repair, p53 a tumor suppressor protein, interacts with single stranded DNA 3’ ends and activate repair process, inhibit CDK-cyclin kinases or it can induce programmed cell-death, called Apoptosis. Inspite of all these checks if the cell acquires the ability to divide and redivide, it leads to cell proliferation without control, that is tumorigenesis.
2 Cells, only when stimulated by growth factors (or sufficient nutrients as in yeast cells), go into cell division mode. Almost all cells do contain variety cell surface receptors for variety of stimuli. When mitogens bind to its specific cell surface receptor, ligand binding activates the receptor. Ligand binding induces conformational changes in the trans-membrane domain of the receptor and it dimerizes at cytosolic side also. If this receptor happens to have Tyrosine kinase activity, it gets activated because of dimerization. This leads to autophosphorylation, thus the membrane receptor becomes fully active. Such activated receptor kinases interact with another protein at cytoplasmic surface, such as G-binding factors and activating them to generate a cascading kinase activity. This can result in phosphorylation of a variety of substrates involved in cell cycle, of which some may be involved in metabolism and growth, some may be s-phase Cdk-Cylins and some may be transcription factors, transcription activators, which on phosphorylation enter the nucleus and bind to their respective DNA sequences and activate genes, whose products are essential for activating more genes that are required for the entry of cell into S-phase and activate DNA replication. Thus cell can enter into cell division cycle.
3 If the gene for cell surface receptor undergoes a mutation where the ligand binding domain is lost, or the cytoplasmic dimerization domain is missing, or a mutation that automatically makes the receptor transmembrane domain becomes dimer, thus the surface receptor protein, though dismembered, remains active even in the absence of the growth hormone or signals or the ligands; this makes the cell to have persisting kinase activity, which has a cascading effect. This can lead to phosphorylation of Cdk-Cyclin inhibitors, thus they release from Cdk-Cyclin from inhibition. RBs are phosphorylated to release all the E2F transcription factors from inhibition. Similarly phosphorylation of Cdc25 activates its phosphatase activity and it activates Cdk-Cyclin kinase by dephosphorylation at Thr 160 (Thr161), thus the MPK becomes active, which can go through a series of activation and inactivation required for repeated cell division. This ends up in unrestricted cell division, leading to cancer. All this is due to one gene and one protein defect.
4 Similarly another transducing kinase, which is found located at the cytosolic side of a receptor kinase receptor, called Ras-kinase, because of its gene, mutations and small deletion at its C-terminal end makes the Ras-kinase to be over active, perhaps ten times the normal Ras enzyme.
5 There are many such kinases, which have important targets, which have implications on regulating the synthesis of factors, which activate transcription of genes required for DNA replication and cytoplasmic division. If such kinases undergo mutation; it can be a point mutation, or deletion, resulting in change in its conformation into ever-active form of kinases. Such kinases with downstream cascading events activate more number of substrates that activate required genes, which can lead to cell proliferation without control.
6 The mutation can be due to translocation of a segment of chromosome containing a gene from its normal position to a site where the gene(s) activated under the influence of its neighbor activator or enhancer sequences thus cause consistent expression and perhaps over expression of a gene placed next to it.
7 Some of the genes may be duplicated, again this cause s production of proteins more than required.
8 Cell cycle events are tightly controlled by a set of genes some of which act as inhibitors at certain check points. Mutation in such genes renders cells incapable of controlled cell cycle. E.g. Suppressor genes such as P53, RB, P16, p21 and others.
9 Mitogens and growth factors induce D-type cyclins in mammalian systems. Cyclins D1, D2 and D3 associate with respective Cdk4 and CdK6 to generate catalytically active kinases.
10 Withdrawal of Mitogens prior to the passage through checkpoints; leads to the accumulation of p21, P27 and P16 proteins. These proteins, especially P16 binds to Cyclin D/CdK4 complex and inhibit cyclin dependent kinase activity; thus cause the arrest at G1 stage
11 Gene amplification or translocation of Cyc-D1 genes under the control of inappropriate promoter leads to over expression of Cyc-D1; this abnormality has been found in many human tumors. In human B-lymphocytes, the cyc-D1 gene is translocated in such a way, its transcription is under the control of an enhancer of antibody producing IgH gene. Thus Cyc-D1 excessively produces even in the absence of mitogens. This is analogous to Myc gene expression in Burkits’ lymphoma, which is also found to be a translocation defective cancer.
12 Over expression of Cyc-D1 under the control of an enhancer in mammary duct of mice has led to the proliferation of ductal cells and the cancer.
13 It is also known now in the case of human breast cancer Cyc-D1 is over expressed.
14 RB is another key protein in cell cycle. In its unphosphorylated state, RB binds to TFs called E2F and prevent E2F mediated transcription of many genes, whose products such as DNA-polymerase and other factors required for DNA replication.
15 P16 by binding to Cyc-D-CdK4 complex makes it as inactive kinase complex. Dissociation of P16 from the complex makes the Cyclin-Cdks complex active. This kinase phosphorylates RB protein and makes it to dissociate form E2F factor.
16 So what makes the P16 to dissociate from cyclin-Cdk protein complex? P16 protein is cyclin-dependent kinase inhibitor. If the P16 undergoes mutation, such as deletion mutation, it cannot bind to cyclin dependent kinase and Cyclin dependent kinase becomes active and phosphorylated RB of the RB-E2F complex thus releases E2F from inhibition.
In mammalian system especially in humans’ loss of P16 function mimics Cyclin-D1 over expression, which leads to hyper phosphorylation of RB protein, thus P16 acts as tumor suppressor.
1 There are examples, when some of the said kinases, though inactive, are translocated to positions, where they are activated by the neighboring sequence domains, such as activators or enhancers of viral LTR sequences or any other gene, which has such activity. This ultimately leads to consistent Kinase activity, which may lead to insistent cell division activity.
2. Cell cycle repressors like RB proteins, its related proteins such as p107 and p130 holds cell cycle by binding to transcription factors from activating genes required for DNA synthesis and other related cell cycle events. Similarly proteins such as p53 an important surveillance factor scans for DNA damage, and if it finds any such damages, it binds to broken ends of DNA and gets activated. The activated p53 in turn activates the expression of genes, which have inhibitory function on many Kinases, thus rescue the cell from going through cell division, if it happens it will have a deleterious effect on cell population. If the damage is severe it activates apoptotic process through the release of cytochrome C from mitochondrial periplasmic space. If mutations in cells disable such genes, cells can easily enter into cell division mode. Majority of the cancers are due to the defects in p53 and it related tumor suppressors (p63 and p73).
2 In some case cellular kinases activate transcriptional factors, which in turn activate genes required for cell division. There are gene products, which are required for differentiation of dividing cells. If one of them mutate, derivatives of cells instead continue to proliferate, which can end up in cancer
3 The above mentioned operative systems and the overall process may start at different sites, by different signals, but ultimately all have converging effect to make cells to be as proliferate as possible and to be eternal.
4 Many viruses induce cancer and the genes are all viral genes. And most of them are DNA viruses. Some of these viruses, on infection code for proteins, which inactivate tumor suppressor gene product p53, e.g. SV40 and other Polyoma viruses and some, activate cellular genes leading to oncogenic pathway. Human Papilloma virus, Adeno virus, Epstein Barr virus and many others cause cancer, again by expressing kinase genes or suppressing tumor suppressor genes.
5 There are many viruses which carry altered cellular genes. Most of them belong to retroviruses. In the case of Rous Sarcoma Virus (RSV), it carries altered cellular Src gene. On infection viral promoter cum enhancers transcribes the Src gene. As the Src gene codes for a kinase, which is produced constitutively; it can lead to oncogenisis. Many retroviruses such as HTLV, HIV, MMTV and others carry defective cellular genes and cause cancer in their hosts.
Therapies to Cancer:
· Hormone therapies- slow or stop the growth of hormone-sensitive tumors, which require certain hormones to grow. Hormone therapies act by preventing the body from producing the hormones or by interfering with the action of the hormones. Hormone therapies have been approved for both breast cancer and prostate cancer.
· Signal transduction inhibitors block the activities of molecules that participate in signal transduction, the process by which a cell responds to signals from its environment. During this process, once a cell has received a specific signal, the signal is relayed within the cell through a series of biochemical reactions that ultimately produce the appropriate response(s). In some cancers, the malignant cells are stimulated to divide continuously without being prompted to do so by external growth factors. Signal transduction inhibitors interfere with this inappropriate signaling.
· Gene expression modulators modify the function of proteins that play a role in controlling gene expression.
· Apoptosis inducers cause cancer cells to undergo a process of controlled cell death called apoptosis. Apoptosis is one method the body uses to get rid of unneeded or abnormal cells, but cancer cells have strategies to avoid apoptosis. Apoptosis inducers can get around these strategies to cause the death of cancer cells.
· Angiogenesis inhibitors block the growth of new blood vessels to tumors (a process called tumor angiogenesis). A blood supply is necessary for tumors to grow beyond a certain size because blood provides the oxygen and nutrients that tumors need for continued growth. Treatments that interfere with angiogenesis may block tumor growth. Some targeted therapies that inhibit angiogenesis interfere with the action of vascular endothelial growth factor (VEGF), a substance that stimulates new blood vessel formation. Other angiogenesis inhibitors target other molecules that stimulate new blood vessel growth.
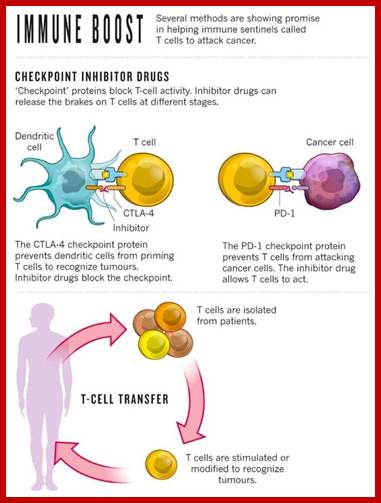
Use of check point inhibitors:
Cells produce several protein inhibitors one such proteins are check point inhibitor proteins. Cell cycle event is regulated by check point proteins. They regulate transition from Go to G1 and G1 to S and G1- to G2 and last is M stage. These are regulated by check points of cell cycle. Use of drugs and antibodies to specific type of check point proteins is immense use in treating cancer.
http://www.vox.com/
Immunotherapies;
Immuno therapy 2016 latest; Immunotherapy triggers the immune system against the said cancer cells to destroy them. Some immunotherapies are monoclonal antibodies that recognize specific molecules on the surface of cancer cells. Binding of the monoclonal antibodies to the target molecules results in the immune based destruction of cells that express target molecule. Other monoclonal antibodies bind to certain immune cells to help these cells better kill cancer cells. Cancer, Immunotherapy CAR T cell immunotherapy works by harvesting a patient’s own T cells, genetically modifying them to improve their ability to identify and destroy harmful cancer cells, and then putting them back in the patient; http://www.cell-sci.com

Researcher Chiara Bonini said: ‘This really is a revolution”.
The treatment is created from T-cells – white blood cells that normally fight off viruses and bacteria – which are removed from the patient and genetically tweaked to recognise and attack their cancer.
The genetically-modified patient’s cells are then grown in their millions in a lab before being infused back into the patient, where they hunt down and destroy the cancer cells.
Scientists around the world are perfecting this technique, and a series of trials have shown it to have remarkable potential.
Some of the famous people missed this therapy and some got cured.
![]()

Fewer than one in five pancreatic cancer patients are given a blood test for elevated levels of a tumor marker can indicate the seriousness of pancreatic cancer and help doctors determine treatment options with better outcomes. Apple CEO Steve Jobs, pictured, died in 2011 due to pancreatic cancer. Historically, only about 7 percent of pancreatic cancer patients have lived at least five years after diagnosis. I-phone discoverer died; File photo by Terry Schmitt/UPI
| License Photo http://www.upi.com/

Jimmy Carter, a cancer patient cured; http://drbarronlerner.com/

Jimmy Carter identified he has cancer melanoma in 1915, now dec.2015, no longer needs Cancer Treatment of Melanoma after he was treated with immunotherapy drug “Pembrolizumab” which worked as check point inhibitor- therapy; former President of USA; http://www.fox29.com/

Little Layla Richards
Little Layla Richards above was given a T-cell treatment so experimental it had only been tested on mice.
The baby, from north London, had one of the worst cases of leukaemia her doctors had seen, and when all other therapies failed, her parents were told to expect the worst.
But they refused to give up and, last summer, when Layla was one, doctors at Great Ormond Street Hospital gave her an infusion of 50million cells genetically engineered to hunt and kill her cancer.
Her mother Lisa Foley, 27, said: ‘We didn’t want to accept palliative care and so we asked doctors to try anything for our daughter, even if it hadn’t been tried before.’
Father Ashleigh Richards, 30, added: ‘It was scary to think the treatment had never been used in a human before but even with the risks there was no doubt we wanted to try.’
The treatment wiped out Layla’s cancer. It is too early to say she is cured, but doctors have described her recovery as a ‘near miracle’. Little Layla Richards was given a T-cell treatment so experimental it had only been tested on mice.
The treatment wiped out Layla’s cancer. It is too early to say she is cured, but doctors have described her recovery as a ‘near miracle’.
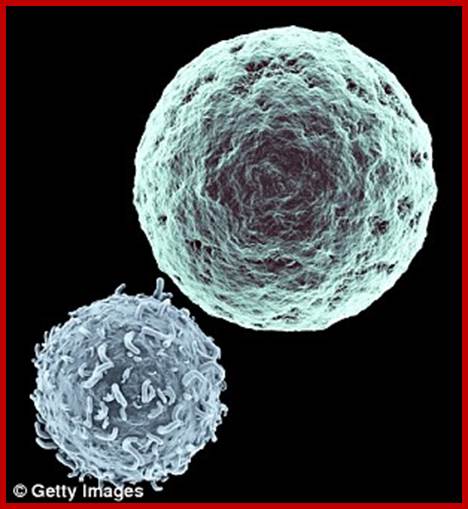

T-cell immunotherapy is created from T-cells bottom left in left image – white blood cells that normally fight off viruses and bacteria. These are removed from the patient and genetically tweaked to recognise and attack their cancer. The genetically-modified cells are then grown in their millions in a lab before being infused back into the patient, where they hunt down cancer cells with specific antigens on cancer cell surface (right).
When scientists at the Fred Hutchinson Cancer Research Centre in Seattle gave genetically-modified T-cells to leukaemia patients with months to live, the cancer disappeared in 94 per cent of cases. Patients with other blood cancers saw response rates of greater than 80 per cent, with more than half experiencing complete remission.
Immunotherapy called adoptive cell transfer (ACT).was/is applied in advanced cancer patients suffering from blood cancer and they are completely recovered. When a patient suffers from cancer, his tumour cells are infiltered with lymphocytes (TILs), they are collected from the patients. The cells are then activated by treating them with activated by treating them with signalling proteins like Cytokines and infused back into patients’ blood stream. The TILs have great perception to recognize and target immune cells and attack them and destroy them. Another alternative therapy is CAR T-cell therapy. In this cells are collected from patients and genetically modified to express a protein known as Chimeric Antigen Receptor (CAR). Such modified cells are grown in the lab into a large population, which are then infused into the patient, the modified T-cells attack cancer cells.
Yet the most challenging aspect is which cancer cell proteins are the best targets for immunotherapy
· Monoclonal antibodies that deliver toxic molecules can cause the death of cancer cells specifically. Once the antibody has bound to its target cell, the toxic molecule that is linked to the antibody—such as a radioactive substance or a poisonous chemical—is taken up by the cell, ultimately killing that cell. The toxin will not affect cells that lack the target for the antibody—i.e., the vast majority of cells in the body.

ds DNA picture used in Chimeric Antigen Receptor Therapies (CAR-T)

Engineered T lymphocytes to target and eradicate malignancy has begun to be realized recently, with remarkable clinical success reported by a number of groups using Chimeric Antigen Receptor –engineered T cells to target CD19-positive hematologic malignancies. Michael Kalo http://jitc.biomedcentral.com/ Molecular engineering and expression of recombinant tumor-specific Chimeric Antigen Receptors (CAR) have shown remarkable promise in clinical trials that target hematological malignancies. CD19+ cells.
Cancer vaccines and gene therapy are sometimes considered targeted therapies because they interfere with the growth of specific cancer cells. Information about these treatments can be found in the NCI fact sheets Cancer Vaccines and Biological Therapies for Cancer.
The most common side effects seen with targeted therapies are diarrhea and liver problems, such as hepatitis and elevated liver enzymes. Other side effects seen with targeted therapies include:
Use of check point inhibitors;

Cells produce several protein inhibitors one such proteins are check point inhibitor proteins. Cell cycle event is regulated by check point proteins. They regulate transition from Go to G1 and G1 to S and G1- to G2 and last is M stage. These are regulated by check point poteins of cell cycle. Use of drugs and antibodies to specific type of check point proteins is immense use in treating cancer.
· What targeted therapies have been approved for specific types of cancer?
· The FDA has approved targeted therapies for the treatment of some patients with the following types of cancer (some targeted therapies have been approved to treat more than one type of cancer):
· Adenocarcinoma of the stomach or gastroesophageal junction: Trastuzumab (Herceptin®), ramucirumab (Cyramza®)
· Basal cell carcinoma: Vismodegib (Erivedge®), sonidegib (Odomzo®)
· Brain cancer: Bevacizumab (Avastin®), everolimus (Afinitor®)
· Breast cancer: Everolimus (Afinitor®), tamoxifen (Nolvadex), toremifene (Fareston®),Trastuzumab (Herceptin®), fulvestrant (Faslodex®), anastrozole (Arimidex®),exemestane (Aromasin®), lapatinib (Tykerb®), letrozole (Femara®), pertuzumab (Perjeta®), ado-trastuzumab emtansine (Kadcyla®), palbociclib (Ibrance®)
· Cervical cancer: Bevacizumab (Avastin®)
· Colorectal cancer: Cetuximab (Erbitux®), panitumumab (Vectibix®), bevacizumab (Avastin®), ziv-aflibercept (Zaltrap®), regorafenib (Stivarga®), ramucirumab (Cyramza®)
· Dermatofibrosarcoma protuberans: Imatinib mesylate (Gleevec®)
· Endocrine/neuroendocrine tumors: Lanreotide acetate (Somatuline® Depot)
· Head and neck cancer: Cetuximab (Erbitux®)
· Gastrointestinal stromal tumor: Imatinib mesylate (Gleevec®), sunitinib (Sutent®),regorafenib (Stivarga®)
· Giant cell tumor of the bone: Denosumab (Xgeva®)
· Kaposi sarcoma: Alitretinoin (Panretin®)
· Kidney cancer: Bevacizumab (Avastin®), sorafenib (Nexavar®), sunitinib (Sutent®),pazopanib (Votrient®), temsirolimus (Torisel®), everolimus (Afinitor®), axitinib (Inlyta®),nivolumab (Opdivo®)
· Leukemia: Tretinoin (Vesanoid®), imatinib mesylate (Gleevec®), dasatinib (Sprycel®),nilotinib (Tasigna®), bosutinib (Bosulif®), rituximab (Rituxan®), alemtuzumab (Campath®), ofatumumab (Arzerra®), obinutuzumab (Gazyva®), ibrutinib (Imbruvica®),idelalisib (Zydelig®), blinatumomab (Blincyto®)
· Liver cancer: Sorafenib (Nexavar®)
· Lung cancer: Bevacizumab (Avastin®), crizotinib (Xalkori®), erlotinib (Tarceva®),gefitinib (Iressa®), afatinib dimaleate (Gilotrif®), ceritinib (LDK378/Zykadia™),ramucirumab (Cyramza®), nivolumab (Opdivo®), pembrolizumab (Keytruda®),osimertinib (Tagrisso™), necitumumab (Portrazza™), alectinib (Alecensa®)
· Lymphoma: Ibritumomab tiuxetan (Zevalin®), denileukin diftitox (Ontak®), brentuximab vedotin (Adcetris®), rituximab (Rituxan®), vorinostat (Zolinza®), romidepsin (Istodax®),bexarotene (Targretin®), bortezomib (Velcade®), pralatrexate (Folotyn®), ibrutinib (Imbruvica®), siltuximab (Sylvant®), idelalisib (Zydelig®), belinostat (Beleodaq®)
· Melanoma: Ipilimumab (Yervoy®), vemurafenib (Zelboraf®), trametinib (Mekinist®),dabrafenib (Tafinlar®), pembrolizumab (Keytruda®), nivolumab (Opdivo®), cobimetinib (Cotellic™)
· Multiple myeloma: Bortezomib (Velcade®), carfilzomib (Kyprolis®), panobinostat (Farydak®), daratumumab (Darzalex™), ixazomib citrate (Ninlaro®), elotuzumab (Empliciti™)
· Myelodysplastic/myeloproliferative disorders: Imatinib mesylate (Gleevec®),ruxolitinib phosphate (Jakafi®)
· Neuroblastoma: Dinutuximab (Unituxin™)
· Ovarian epithelial/fallopian tube/primary peritoneal cancers: Bevacizumab (Avastin®), olaparib (Lynparza™)
· Pancreatic cancer: Erlotinib (Tarceva®), everolimus (Afinitor®), sunitinib (Sutent®)
· Prostate cancer: Cabazitaxel (Jevtana®), enzalutamide (Xtandi®), abiraterone acetate (Zytiga®), radium 223 dichloride (Xofigo®)
· Soft tissue sarcoma: Pazopanib (Votrient®)
· Systemic mastocytosis: Imatinib mesylate (Gleevec®)
· Thyroid cancer: Cabozantinib (Cometriq®), vandetanib (Caprelsa®), sorafenib (Nexavar®), lenvatinib mesylate (Lenvima®)
· Signal transduction inhibitors
Apoptosis Inducers: Angiogenesis Inhibitors immunotherapy; Monoclonal antibody conjugate therapy; http://www.healthline.com/Programmed Cell Death:

TCR- and cytokine-driven activation of iNKT cells; Two signals are involved in the physiological activation of invariant natural killer T (iNKT) cells: a T cell receptor (TCR) signal provided by a lipid–CD1d complex; and a cytokine signal that depends on the constitutive expression of certain cytokine receptors by iNKT cells. The left panel shows activation by a strong foreign antigen, which is dominated by the TCR signal and has little dependence on antigen-presenting cell (APC)-derived cytokines that are generated in response to the stimulation of pattern-recognition receptors (PRRs). By contrast, the right panel shows cytokine-dominated iNKT cell activation. In this scenario, PRR-mediated activation of APCs leads to the generation of pro-inflammatory cytokines such as interleukin-12 (IL-12). For cytokine-mediated activation, a TCR signal is still required in most cases and can be provided by a low-affinity microbial lipid antigen or self lipid antigen. The relative contributions of TCR and cytokine signals to iNKT cell activation are likely to be context dependent. Patrick J. Brennan, Manfred Brigl & Michael B. Brenner; ;http://www.nature.com
Invariant natural killer T cells: An innate activation scheme linked to diverse effector
functions;
two
signals are involved in the physiological activation of invariant natural
killer T (iNKT) cells: a T cell receptor (TCR) signal provided by a lipid–CD1d
complex; and a cytokine signal that depends on the constitutive expression of
certain cytokine receptors by iNKT cells. The left panel shows activation by a
strong foreign antigen, which is dominated by the TCR signal and has little
dependence on antigen-presenting cell (APC)-derived cytokines that are
generated in response to the stimulation of pattern-recognition receptors
(PRRs). By contrast, the right panel shows cytokine-dominated iNKT cell
activation. In this scenario, PRR-mediated activation of APCs leads to the
generation of pro-inflammatory cytokines such as interleukin-12 (IL-12). For
cytokine-mediated activation, a TCR signal is still required in most cases and
can be provided by a low-affinity microbial lipid antigen or self-lipid
antigen. The relative contributions of TCR and cytokine signals to iNKT cell
activation are likely to be context dependent. Two signals are involved in the
physiological activation of invariant natural killer T (iNKT) cells: a T cell
receptor (TCR) signal provided by a lipid–CD1d complex; and a cytokine signal
that depends on the constitutive expression of certain cytokine receptors by
iNKT cells. The left panel shows activation by a strong foreign antigen, which
is dominated by the TCR signal and has little dependence on antigen-presenting
cell (APC)-derived cytokines that are generated in response to the stimulation
of pattern-recognition receptors (PRRs). By contrast, the right panel shows
cytokine-dominated iNKT cell activation. In this scenario, PRR-mediated activation
of APCs leads to the generation of pro-inflammatory cytokines such as
interleukin-12 (IL-12). For cytokine-mediated activation, a TCR signal is still
required in most cases and can be provided by a low-affinity microbial lipid
antigen or self-lipid antigen. The relative contributions of TCR and cytokine
signals to iNKT cell activation are likely to be context dependent. Nature
Reviews; immunology;
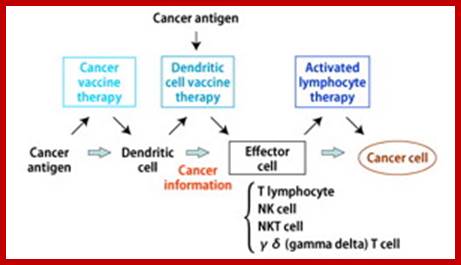
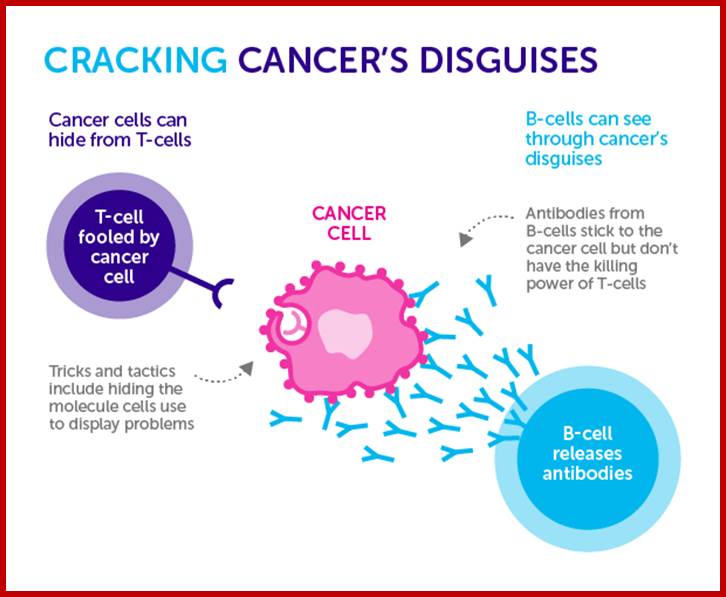
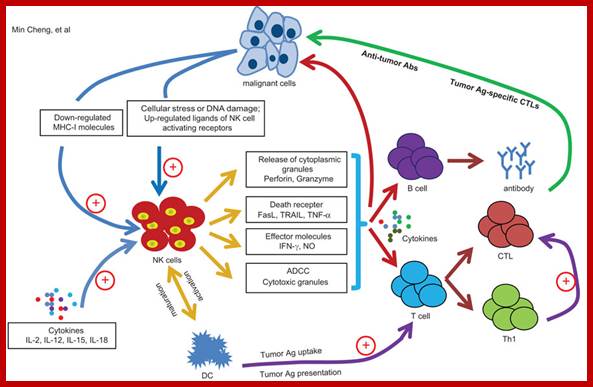
In cancer immunotherapy, the process of immune reaction to cancer is effectively used for treatment. Immunotherapy is roughly divided into cancer vaccine therapy, dendritic cell therapy, and activated lymphocyte therapy. Furthermore, activated lymphocyte therapy includes T lymphocyte (T cell) therapy, NK (natural killer) cell therapy, NKT cell therapy, and γδT cell therapy. http://www.nco-clinic.jp/e
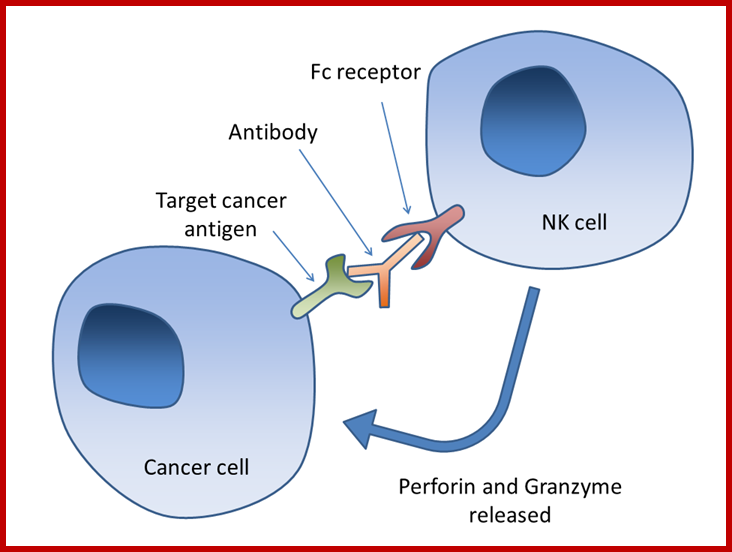
Antibody-dependent cell-mediated cytotoxicity:::

Fc regions of antibodies bound to cancer cells, the NK cell releases perforin and granzyme, leading to cancer cell apoptosis; Chimaeric Antigen Receptor T-cells, or CAR T-cells Wikipedia.org; http://scienceblog.cancerresearchuk.org/2016-feb
Targeted therapies like imatinib (Gleevec®) and trastuzumab (Herceptin®)—drugs that target cancer cells by homing in on specific molecular changes seen primarily in those cells—http://www.cancer.gov/
The T-cell treatment works best with blood cancers rather than tumors.
Treatment Types
Find out what you need to know about the most common types of cancer treatment, such as surgery, chemotherapy, radiation therapy, and many others. Learn how they work and why they are used, and get an idea of what to expect and how they might affect you if you're getting them.
Surgery
Surgery can be used to diagnose, treat, or even help prevent cancer in some cases. Most people with cancer will have some type of surgery. It often offers the greatest chance for cure, especially if the cancer has not spread to other parts of the body. Learn more about surgery here.
Chemotherapy
Chemotherapy (chemo) is the use of medicines or drugs to treat cancer. The thought of having chemotherapy frightens many people. But knowing what chemotherapy is, how it works, and what to expect can often help calm your fears. It can also give you a better sense of control over your cancer treatment.
Radiation Therapy
Radiation therapy uses high-energy particles or waves to destroy or damage cancer cells. It is one of the most common treatments for cancer, either by itself or along with other forms of treatment. Learn more about radiation therapy in this section.
Targeted Therapy
Targeted therapy is a newer type of cancer treatment that uses drugs or other substances to more precisely identify and attack cancer cells, usually while doing little damage to normal cells. Targeted therapy is a growing part of many cancer treatment regimens. Find out more about it here.
Immunotherapy
Immunotherapy is treatment that uses your body's own immune system to help fight cancer. Get information about the different types of immunotherapy and the types of cancer they are used to treat.
Hyperthermia
The idea of using heat to treat cancer has been around for some time, but early attempts had mixed results. Today, newer tools allow more precise delivery of heat, and hyperthermia is being studied for use against many types of cancer.
Stem Cell Transplant (Peripheral Blood, Bone Marrow, and Cord Blood Transplants)
Here we offer a review of bone marrow transplants and other types of stem cell transplants that are used to treat cancer. We outline what a transplant is like for most people, and discuss some of the issues that come with it.
Photodynamic Therapy
Photodynamic therapy or PDT is a treatment that uses special drugs, called photosensitizing agents, along with light to kill cancer cells. The drugs only work after they have been activated or "turned on" by certain kinds of light.
Lasers in Cancer Treatment
Lasers, which are very powerful, precise beams of light, can be used instead of blades (scalpels) for very careful surgical work, including treating some cancers.
Blood Product Donation and Transfusion
Transfusions of blood and blood products temporarily replace parts of the blood when a person's body can't make its own or has lost them from bleeding. Here, we describe blood and its components and why they are important. We also explain how blood is donated and transfused and how this relates to people with cancer.
I Can Cope Online Class - Understanding Cancer Treatments
For a quick, easy way to learn important facts and practical tips about cancer and related issues, participate in our interactive online program, I Can Cope—Online. There is never any charge to participate, and you set the pace—whenever and wherever is most convenient for you. This class focuses on understanding cancer treatments.
Number of Cases, Deaths, and Survivors
Worldwide in 2012 (the latest year for which information is available)—
· 14.1 million new cancer cases were diagnosed.
· 8.2 million people died from cancer.
· 32.6 million people were five-year cancer survivors (people who are alive five years after being diagnosed with cancer).
By 2025, 19.3 million new cancer cases are expected to be diagnosed each year.
In less developed regions in 2012—
· 8 million new cancer cases were diagnosed (57% of the global total).
· 5.3 million people died from cancer (65% of the global total).
· 15.6 million people were five-year cancer survivors (48% of the global total).
- Cancer: Facts, Causes, Symptoms and Research

Cancer Cases
In 2012, the most common cancers worldwide (for both sexes) were*—
1. Lung cancer (13% of all cancers diagnosed; 1.8 million people).
2. Breast cancer (12% of all cancers diagnosed; 1.7 million people).
3. Colorectal cancer (10% of all cancers diagnosed; 1.4 million people).
4. Prostate cancer (8% of all cancers diagnosed; 1.1 million people).
5. Stomach cancer (7% of all cancers diagnosed; 952,000 people).
6. Liver cancer (6% of all cancers diagnosed; 782,000 people).
7. Cervical cancer (4% of all cancers diagnosed; 528,000 people).
In 2012, the most commonly diagnosed cancers worldwide (for males and females) were—
· Among males: Lung, prostate, colorectal, stomach, and liver.
· Among females: Breast, colorectal, lung, cervical, and stomach.

Cancer Deaths
An estimated 168.1 million years of healthy life are lost due to cancer every year.
In 2012, the most common causes of cancer death worldwide (for both sexes) were*—
1. Lung cancer (19% of all cancer deaths; 1.6 million people).
2. Liver cancer (9% of all cancer deaths; 745,000 people).
3. Stomach cancer (9% of all cancer deaths; 723,000 people).
4. Colorectal cancer (9% of all cancer deaths; 694,000 people).
5. Breast cancer (6% of all cancer deaths; 522,000 people).
6. Cancer of the esophagus (5% of all cancers diagnosed; 400,000 people).
7. Pancreas cancer (4% of all cancers diagnosed; 330,000 people).
In 2012, the most common causes of cancer death worldwide (for males and females) were—
· Among males: Lung, liver, stomach, colorectal, and prostate.
· Among females: Breast, lung, colorectal, cervical, and stomach.
*Note: Rankings are defined by the total number of cases and deaths and are not age-standardized.
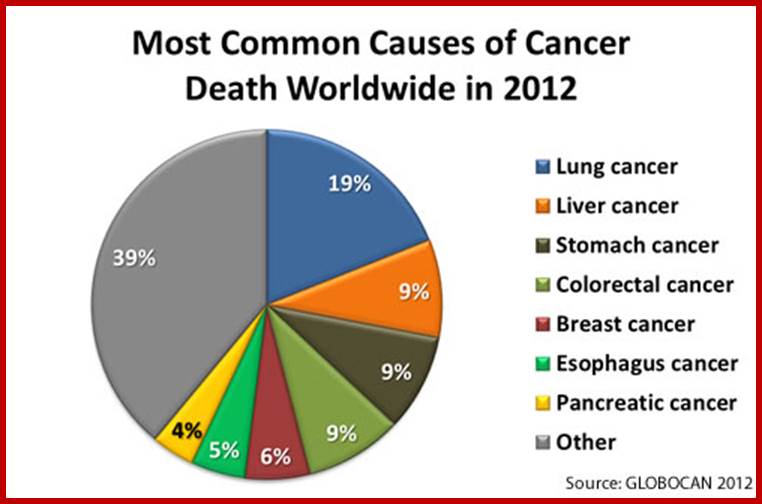
Data source: GLOBOCAN 2012: Estimated Cancer Incidence, Mortality and Prevalence Worldwide in 2012
T cell targets for immunoregulatory antibody therapy. Immunotherapy-2016Feb: http://www.nature.com/
Cancer immunotherapy comes of Ages;


http://scienceblog.cancerresearchuk.org/
http://scienceblog.cancerresearchuk.org/

http://scienceblog.cancerresearchuk.org/
Nature reviews: Immunology
In addition to specific antigen recognition through the TCR, T-cell activation is regulated through a balance of positive and negative signals provided by co-stimulatory receptors. These surface proteins are typically members of either the TNF receptor or B7 super families. Agonistic antibodies directed against activating co-stimulatory molecules and blocking antibodies against negative co-stimulatory molecules may enhance T-cell stimulation to promote tumor destruction

Nature reviews: Immunology
Biological activities of CTLA-4 antibody blockade. a. On encountering a dendritic cell presenting a cognate tumour-antigen-derived peptide epitope and expressing B7 co-stimulatory molecules (CD80, CD86), specific antitumour T cells become activated through TCR and CD28 signalling. b, CTLA-4 is subsequently upregulated and preferentially engages B7 to attenuate T-cell response. c, Ipilimumab blocks CTLA-4 function, thereby allowing enhanced T-cell stimulation and a more potent antitumour reaction. Ipilimumab may also antagonize CTLA-4 on regulatory T cells to limit their ability to suppress the antitumour T-cell effector response (not shown). d, CTLA-4 antibody blockade compromises tolerance to some normal tissue antigens, provoking inflammatory toxicities that can have an impact on the skin, pituitary gland and intestine in human patients.
http://www.cancer.gov
What is adoptive T-cell transfer therapy?
Adoptive cell transfer is an experimental anticancer therapy that attempts to enhance the natural cancer-fighting ability of a patient’s T cells. In one form of this therapy, researchers first harvest cytotoxic T cells that have invaded a patient’s tumor. They then identify the cells with the greatest antitumor activity and grow large populations of those cells in a laboratory. The patients are then treated to deplete their immune cells, and the laboratory-grown T cells are infused into the patients.
In another, more recently developed form of this therapy, which is also a kind of gene therapy, researchers isolate T cells from a small sample of the patient’s blood. They genetically modify the cells by inserting the gene for a receptor that recognizes an antigen specific to the patient’s cancer cells and grow large numbers of these modified cells in culture. The genetically modified cells are then infused into patients whose immune cells have been depleted. The receptor expressed by the modified T cells allows these cells to attach to antigens on the surface of the tumor cells, which activates the T cells to attack and kill the tumor cells.
Adoptive T-cell transfer was first studied for the treatment of metastatic melanoma because melanomas often cause a substantial immune response, with many tumor-invading cytotoxic T cells. Adoptive cell transfer with genetically modified T cells is also being investigated as a treatment for other solid tumors, as well as for hematologic cancers.
What are monoclonal antibodies, and how are they used in cancer treatment?
Monoclonal antibodies, or MAbs, are laboratory-produced antibodies that bind to specific antigens expressed by cells, such as a protein that is present on the surface of cancer cells but is absent from (or expressed at lower levels by) normal cells.
To create MAbs, researchers inject mice with an antigen from human cells. They then harvest the antibody-producing cells from the mice and individually fuse them with a myeloma cell (cancerous B cell) to produce a fusion cell known as a hybridoma. Each hybridoma then divides to produce identical daughter cells or clones—hence the term “monoclonal”—and antibodies secreted by different clones are tested to identify the antibodies that bind most strongly to the antigen. Large quantities of antibodies can be produced by these immortal hybridoma cells. Because mouse antibodies can themselves elicit an immune response in humans, which would reduce their effectiveness, mouse antibodies are often “humanized” by replacing as much of the mouse portion of the antibody as possible with human portions. This is done through genetic engineering.
Some MAbs stimulate an immune response that destroys cancer cells. Similar to the antibodies produced naturally by B cells, these MAbs “coat” the cancer cell surface, triggering its destruction by the immune system. FDA-approved MAbs of this type include rituximab, which targets the CD20 antigen found on non-Hodgkin lymphoma cells, and alemtuzumab, which targets the CD52 antigen found on B-cell chronic lymphocytic leukemia (CLL) cells. Rituximab may also trigger cell death (apoptosis) directly.
Another group of MAbs stimulates an anticancer immune response by binding to receptors on the surface of immune cells and inhibiting signals that prevent immune cells from attacking the body’s own tissues, including cancer cells. One such MAb, ipilimumab, has been approved by the FDA for treatment of metastaticmelanoma, and others are being investigated in clinical studies (2).
Other MAbs interfere with the action of proteins that are necessary for tumor growth. For example, bevacizumab targets vascular endothelial growth factor (VEGF), a protein secreted by tumor cells and other cells in the tumor’s microenvironment that promotes the development of tumor blood vessels. When bound to bevacizumab, VEGF cannot interact with its cellular receptor, preventing the signaling that leads to the growth of new blood vessels.
Similarly, cetuximab and panitumumab target the epidermal growth factor receptor (EGFR), and trastuzumabtargets the human epidermal growth factor receptor 2 (HER-2). MAbs that bind to cell surface growth factorreceptors prevent the targeted receptors from sending their normal growth-promoting signals. They may also trigger apoptosis and activate the immune system to destroy tumor cells.
Another group of cancer therapeutic MAbs are the immunoconjugates. These MAbs, which are sometimes called immunotoxins or antibody-drug conjugates, consist of an antibody attached to a cell-killing substance, such as a plant or bacterial toxin, a chemotherapy drug, or a radioactive molecule. The antibody latches onto its specific antigen on the surface of a cancer cell, and the cell-killing substance is taken up by the cell. FDA-approved conjugated MAbs that work this way include 90Y-ibritumomab tiuxetan, which targets the CD20 antigen to deliver radioactive yttrium-90 to B-cell non-Hodgkin lymphoma cells, and ado-trastuzumab emtansine, which targets the HER-2 molecule to deliver the drug DM1, which inhibits cell proliferation, to HER-2 expressing metastatic breast cancer cells.
What are cytokines, and how are they used in cancer treatment?
Cytokines are signaling proteins that are produced by white blood cells. They help mediate and regulate immune responses, inflammation, and hematopoiesis (new blood cell formation). Two types of cytokines are used to treat patients with cancer: interferons (INFs) and interleukins (ILs). A third type, called hematopoietic growth factors, is used to counteract some of the side effects of certain chemotherapy regimens.
Researchers have found that one type of INF, INF-alfa, can enhance a patient’s immune response to cancer cells by activating certain white blood cells, such as natural killer cells and dendritic cells (3). INF-alfa may also inhibit the growth of cancer cells or promote their death (4,5). INF-alfa has been approved for the treatment of melanoma, Kaposi sarcoma, and several hematologic cancers.
Like INFs, ILs play important roles in the body’s normal immune response and in the immune system’s ability to respond to cancer. Researchers have identified more than a dozen distinct ILs, including IL-2, which is also called T-cell growth factor. IL-2 is naturally produced by activated T cells. It increases the proliferation of white blood cells, including cytotoxic T cells and natural killer cells, leading to an enhanced anticancer immune response (6). IL-2 also facilitates the production of antibodies by B cells to further target cancer cells. Aldesleukin, IL-2 that is made in a laboratory, has been approved for the treatment of metastatic kidney cancer and metastatic melanoma. Researchers are currently investigating whether combining aldesleukin treatment with other types of biological therapies may enhance its anticancer effects.
Hematopoietic growth factors are a special class of naturally occurring cytokines. All blood cells arise from hematopoietic stem cells in the bone marrow. Because chemotherapy drugs target proliferating cells, including normal blood stem cells, chemotherapy depletes these stem cells and the blood cells that they produce. Loss of red blood cells, which transport oxygen and nutrients throughout the body, can causeanemia. A decrease in platelets, which are responsible for blood clotting, often leads to abnormal bleeding. Finally, lower white blood cell counts leave chemotherapy patients vulnerable to infections.
Several growth factors that promote the growth of these various blood cell populations have been approved for clinical use. Erythropoietin stimulates red blood cell formation, and IL-11 increases platelet production. Granulocyte-macrophage colony-stimulating factor (GM-CSF) and granulocyte colony-stimulating factor (G-CSF) both increase the number of white blood cells, reducing the risk of infections. Treatment with these factors allows patients to continue chemotherapy regimens that might otherwise be stopped temporarily or modified to reduce the drug doses because of low blood cell numbers.
G-CSF and GM-CSF can also enhance the immune system’s specific anticancer responses by increasing the number of cancer-fighting T cells. Thus, GM-CSF and G-CSF are used in combination with other biological therapies to strengthen anticancer immune responses.
Chimeric antigen receptor (CAR) T-cell therapy:
This is a promising new way to get immune cells called T cells to fight cancer. For this technique, T cells are removed from the patient’s blood and genetically altered in the lab to have specific antigen receptors (called chimeric antigen receptors, or CARs) on their surface. These receptors will attach to proteins on the surface of cancer cells. The T cells are then multiplied in the lab and infused back into the patient’s blood, where they can now seek out the cancer cells and launch a precise immune attack against them.
This technique has shown very encouraging results in early clinical trials against some advanced, hard-to-treat types of leukemia and lymphomas. In many people the cancer could no longer be detected after treatment, although it’s not yet clear if these people have been cured.
Some people have had serious side effects from this treatment, including very high fevers and dangerously low blood pressure in the days after it’s given. Doctors are learning how to manage these side effects.
Doctors are still improving how they make the T cells and are learning the best ways to use them. They are also studying whether this treatment will work for other types of cancer. CAR T-cell therapy is only available in clinical trials at this time. http://www.cancer.org/

I had months to live- now my tumours have disappeared; Beware of hype “miracle’ cancer drugs; http://www.telegraph.co.uk/
Cancer therapy using Light:
IISc scientists Dr. Chakravarty in Bangalore used a new bio-molecule Vanadium which carries chloride ion, when used due to it is cross links to DNA; it leads to cell death when chemical reagents react at two different positions in DNA- the treatment called Photodynamic Therapy. In this the chemical used is photosensitive and it is used to kill small group of cells- http://www.cancer.gov