Genetics of Cancer II
Cancer Genes:
In the first part, Cancer-I, a general account of tumorigenesis has been delineated. In the second section, Cancer-II, few individual genes responsible for cancer have been described, how and why the cellular genes cause cancer. Among 291-310 cancer genes few of the genes are involved in initiating tumorigenesis are cellular genes. Many of the cellular genes have the potential, if undergo mutation, to become cancer genes, they are called proto-oncogenes and the fully functional cancer-causing genes are called oncogenes. It is interesting to know that some foreign genes can also transform cells into cancer cells.

This is picture from the cover
Page of the famous journal -CELL
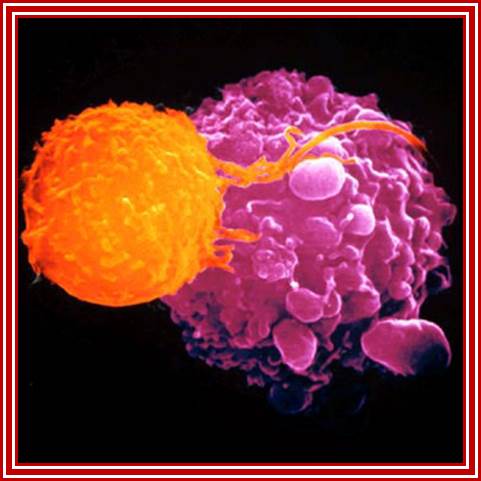
Killer T cells (oranage) are recruited to attack malignant cells in the viral (Herpes) based therapy-T-VEC; This Picture depicts the full blown cancer tissue in metastasis stage; Heide Ledford http://www.nature.com/
Many genetic factors (multifactorial and mutated) are responsible for inducing cancer; they can be secretory factors for signaling pathways, transmembrane receptor (kinases), cellular non-receptor protein kinases, transcription factors and regulators of gene expression. Some RNA or DNA viral genes can also induce cancer and the loss of functioning of cell cycle check point proteins and tumor suppressor genes are the last hurdle and they have been made nonfunctional and the cancer cells pass to become cancerous. Cancerous cells do changes and make many genes cancerous genes. It need not be due to one factor, it is always due to many factors cancers develop. They act one after the other independently and in combination.
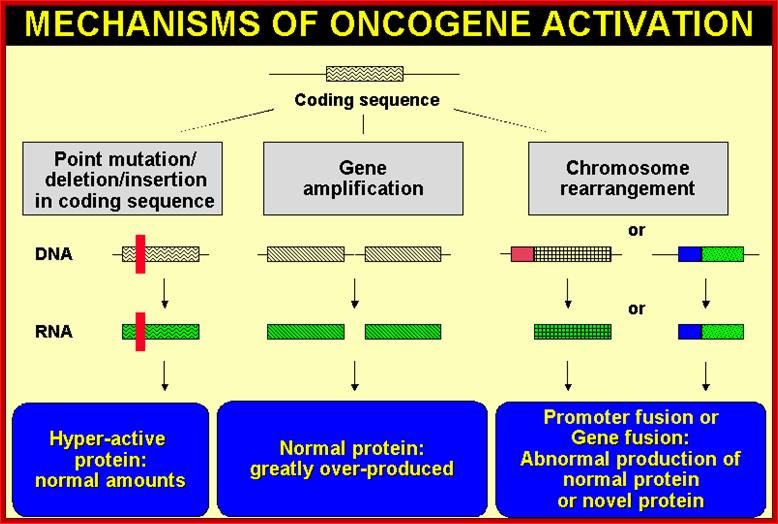
Whatever genes that have been identified as cancer causing genes, they can be 291 to 310 of them (Sangers’), they are all host cell genes suffered some genetic damage in different forms and become nonfunctional or over functional so that they make the cell all the time active, or such genes have been amplified for excess production of the same product, or it can due to non reciprocal translocation where a gene is truncated but allowed expression with the ligand or signals, or they are placed next to an enhancer of the gene that is continuously expressed, or this can be due to the retroviral genome carrying a cellular gene modified is inserted in a region where the modified gene is expressed under the control of retroviral promoter-enhancer regions or this can be due to certain DNA viral infections where the early gene products have a significant effect in terms of activation genes for expression cell cycle events, or they bind to tumor suppressors, that makes the cells to be vulnerable for proliferation or the loss of tumor suppressors, which are the guardians that prevent the cells to become cancers cell by inducing gene expressions to block the cell cycle promoting factors or inducing apoptosis to salvage the cancer from proliferating. Here only few of the well-known genes and their factors involved and the mechanisms involved in cancer development have been described.
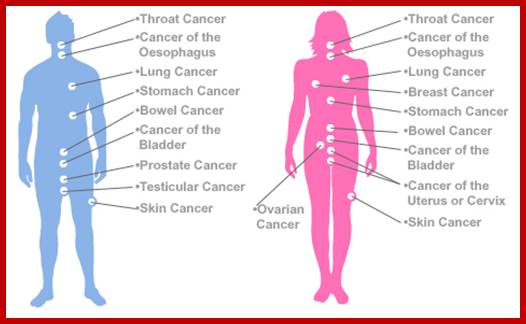
Ten Types of cancers- a general viw; http://www.quantumbooks.com/
Note: There are many other types but shown only few
Oncogenomics: Genomics which shows genes that can initiate or progress or genes fail to stop mitosis of cancer cell development. It gives information on genomic and epigenomic and its transcriptomics of cancer. Oncogenomics also shows identify new oncogenes, or tumour suppressor genes and provide new insights into cancer diagnosis and cancer therapies.
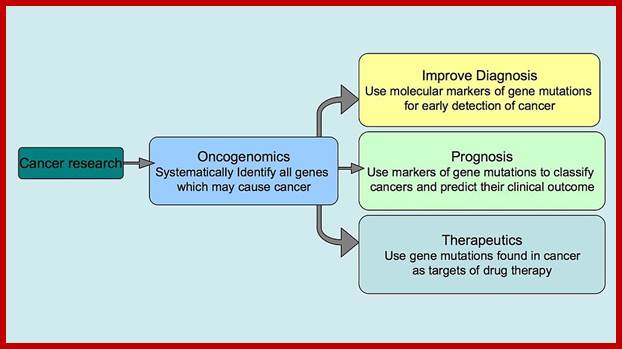
https://en.wikipedia.org
Even mitochondrial DNA has contribution in developing cancer; point mutations, deletions, insertions and copy-number mutations are involved in cancer development. Mitochondrial mutations have been observed in Bladder, Breast, Head and neck, oral, hepatocellular, esophageal, gastric and prostate cancers. Nearly 57.7% (500/867) contained somatic point mutations and of the 1172 mutations surveyed 37.8% (443/1127) were located in the D-loop control region, 13.1% (154/1172) were located in the tRNA or rRNA genes and 49.1% (575/1127) were found in the mRNA genes needed for producing complexes required for mitochondrial respiration. https://en.wikipedia.org
Cancer Causing Genes, Few Examples:
Secreted proteins in signaling pathway:
C-Sis: Class I growth factors: PDFG b-chain.
Ks/HST: Related to FGF (fibroblast growth factors).
Wnt 1: wingless.
KGF (Hst): They encode FGF related growth factors.
Int1 & 2: Related to FGF (fibroblast growth factors) growth factor.
EGF: estrogen growth factor,
ERGF: epidermal growth factor
SFFP: Erythropoietin simulators- a viral glycoprotein.
BFGF; Tumor fibroblast growth factors,
TGF: tissue forming growth factor,
VEGF: Vascular endothelial Growth factor.
SFFV: spleen focus forming virus- that causes erythroleukemia.
HPV : Human papilloma viral (HPV) protein induced caner
Transmembrane Receptor:
Many of them Enzyme linked receptors; Tyrosine kinase and tyrosine kinases associated receptors, Receptor tyrosine phosphatase, receptors serine and threonine kinase, histidine associated kinase.
Tyrosine kinases-receptor bound enzymes.
Erb-B &B2: C-erb.B; Class IIA cell surface receptors: Epidermal Growth PDGFR - receptor protein kinase.
EGFR-receptor tyrosine kinase,
FGFR;
IR:
NGF-
Neu: C-erbB2, 3; EGF like receptor kinase.
Fms: C-fms -> CSF-1 (colony stimulating factor) receptor kinase.
Flg: Fms Like Gene encodes FGF receptors.
Trk (track): Trk-A, B and C are related, encodes the NGF receptor like proteins.
PDFG-platelet derived growth Factor.
FGFR-Fibroblast growth factor,
HGF- encodes hepatocyte growth factor receptor
IGF-1-Insulin like growth factor
VEGF-
M-CSF-
Met: (HGF).
MAPK- mitogen activated kinase
Flg (flag): fms like, encodes a form of FGF receptor.
C-kit: Steel receptor kinase, encodes mast cell growth factor receptor.
C-Moss -> angiotensin receptor.
Her2: human epidermal receptor kinase,
C-mos: angiotensin receptor kinase,
Gs/mitogen-activated protein kinase
TRK: NGF receptor like tyrosine kinase,
GPCR- linked MAPK,MEK,MAP or ERK.
Mitogen activated protein kinase KinaseK,
Mitogenic Signaling of Urokinase Receptor;
Cytosolic-non receptor proteins:
Involved in Signal transduction:
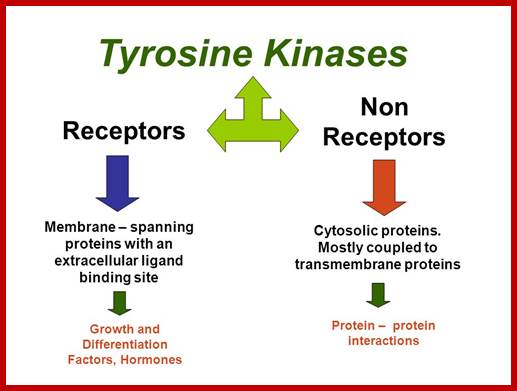
http://slideplayer.com/
Tyrosine kinases:
They are cytoplasmic enzymes (nRTKs), they transfer Phosphate from nucleoside triphosphate donor such as ATP to the target compounds, nRTKs are a subgroup of protein tyrosine kinase family. They regulate many cellular functions. Thirty two such enzymes have been identified in human cells. They regulate cell growth, proliferation, differentiation, migration and apoptosis; they are very important in regulating immune T cells and B cells. Examples-ABL family, ACK family, CSK family, FAK family, FES family, FRK family, JAK family, SRC family TEC family and SYK family.
Non-receptor ( NRTK): they are protein kinases( more than 90 TKs); SRC, ABL, FAK, Janus.
C-Ras->Class IIIC: G binding protein, -membrane associated Tyrosine kinase
Gsp/gip -> Gas & Gai.
LCK:-> from T-cell tumor lines (LYSTRA Cell Kinase)
Mas: It acts as a receptor for Angiotensin, in mammary carcinoma
C-Src-, membrane associated (Class IIIA), (TKs).
C-abl: Class IIIA, Abelson intracellular signal transducers (TrKs).
C-fps:
NRTK: non-receptor tyrosine kinase
Serine-Threonine Kinases:
C-erk: serine Threonine kinase
C-vav:
C-rap:
C-raf: in RTK pathways, phosphorylates Threonine of MAP kinase protein.
C-mos: Class IIIB, intracellular signal transducers (ser/thr kinases)
Nuclear DNA binding proteins –NFs and Transcription factors-TFs:
Transcriptional regulators: activators or co activators.
C-myc: Class IV: Myelocytomatosis, an avian virus HLH protein.
C-Myb: TF.
C-Fos: Class IV: Finkel Osteogenic Sarcoma, Leucine zipper protein.
C-Jun: Class IV: Leucine zipper protein.
C-Rel: NF-kB family of TFs.
C-erb-A: Thyroxine receptor, activator / co activator (Class IIB).
P53- Binds to specific sequence in promoter of DNA.
RB protein; binds to transactivation domain DNA through E2F factors.
DNA Viral genes (Tumor inducers):
Adeno virus (ADV): E1 A and E1B,
Polyoma virus: large T, middle T and small t antigens.
SV 40: T and t antigens
Human Papilloma Virus: E5, E6 and E7 (E2 represses HPV E6 and E7)
Epstein Barr Virus (EBV): EBV 1, -breast cancer
MMTV Env: Mouse mammary tumor virus.
HPV,HBV and MCV.
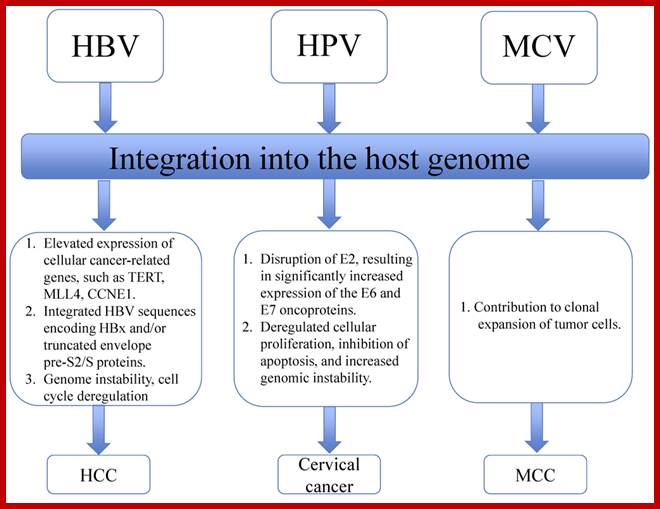
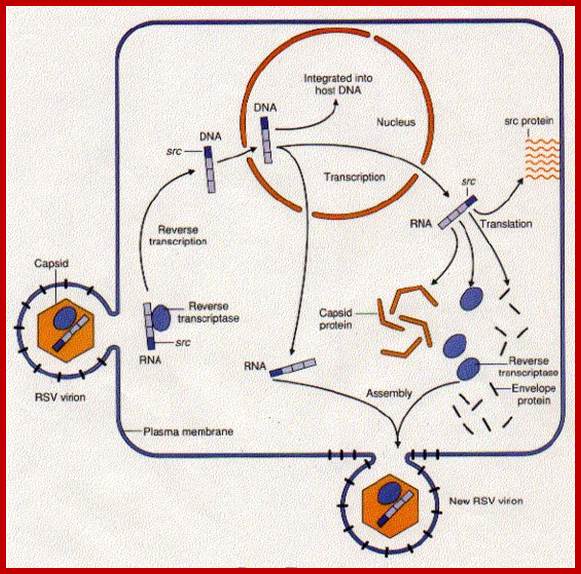
http://www.biology-pages.info/R/RSV.html
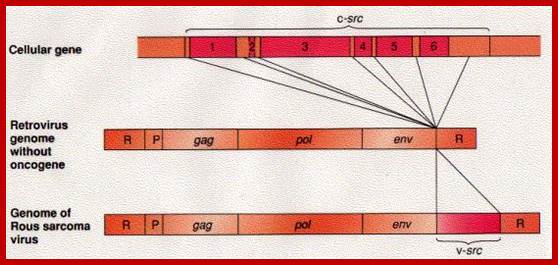
http://www.biology-pages.info/R/RSV.html
Tumor Suppressors Genes:
Class V: Tumor suppressors.
Retinoblastoma (RB),
RB related proteins: p107, p130,
P53 proteins,
P63,
P73,
APC1: Adenomatasis polyposis coli.
FAPC: Familial Adenomatous polyposis colorectal cancer; signaling through adhesion-molecules to nuclear, tumor suppressor.
BRAC 1 and 2: Breast cancer.
Wnt1: wingless, involved in transcriptional regulation.
Wt: Wilms tumor protein- a tumor suppressor,
Many cell cyclin Cdk/Cyclins-inhibitors.
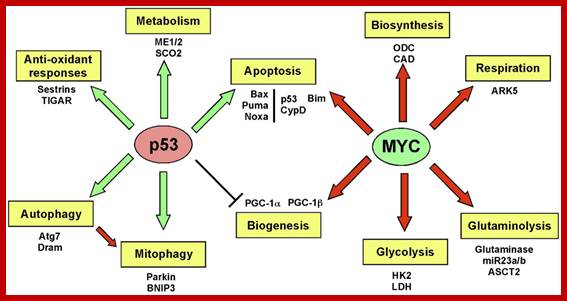
Oncogene (Myc) and tumour suppressor gene (P53) regulation of cell cycle and cellular and Nuclear DNA damage; http://journal.frontiersin.org/
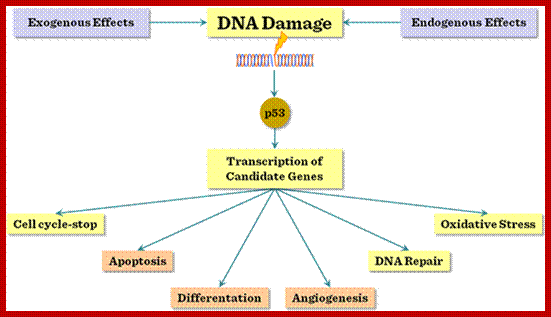
Certain mammalian species of relatively large body mass, including humans, are long-lived. For this purpose, mammalians harbor stem cells in order to clonally replenish tissues. As a result extended mean life-span is attributed with the increased incidence of cancer development, partly due to the prolonged exposure of clonally expanding stem cells to mutagenic factors, compared to short-lived animals almost exclusively composed of post-mitotic cells (i.e. insects). Tumor development is efficiently halted by tumor suppressor genes. This chapter will discuss the ambivalent role of certain tumor suppressor.
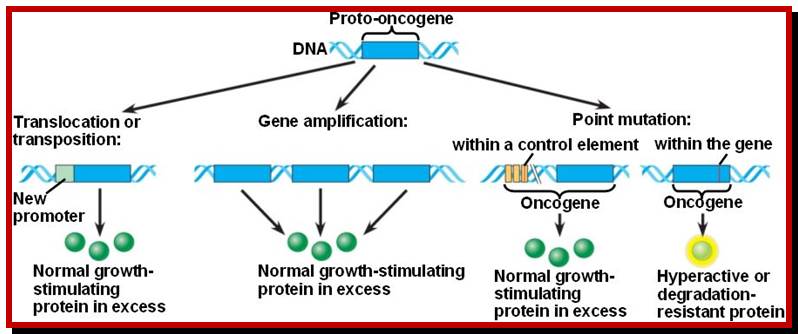
Proto-oncogenes, which are normal and functional cellular genes, code for secreted proteins, transmembrane proteins, cytoplsmic proteins or nuclear proteins; all are cellular genes, but some have potency to induce cancer or suppress cancer. They have the potentiality to become oncogenes provided if they suffer some damage in their gene’ structure and function, so the damage (mutation) leads to the gain of function for cancer causing genes.
Note that all oncogenes, directly or indirectly, are connected to cell receptors activated and induced cell cycle events. Oncogene induced transformed cells lack regulation and restriction imposed on normal cell’s cell division cycle. This is because the cellular proto-oncogenes are mutated and express so to promote uncontrolled cell division that induces cancer. Check point proteins not working. Few factors and their genes responsible for initiating or inducing cancer are discussed briefly.
Signaling Molecules as Cancer Regulating Components:
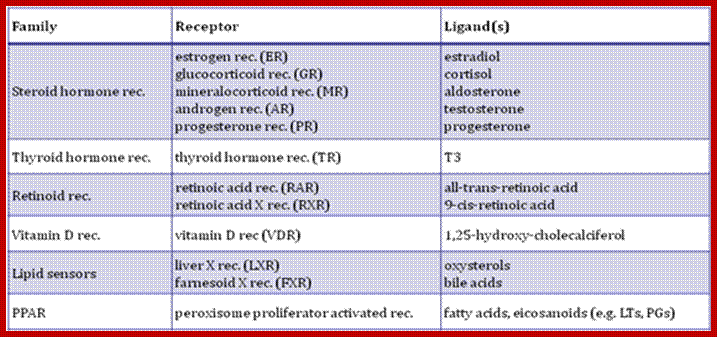
The receptors found in cells are of different types; Cell surface receptors, nuclear receptor families and Intracellular receptor families. Once signals are transduced activated nuclear factors, they act on nuclear receptors
Nuclear receptor families have six domains. The N-terminal A/B domain, central domain and DNA binding domain and ligand binding domain; Activation function (AF)-1/2 sequences are found in the N-/C-terminal domains, with ligand-dependent or –independent regulatory functions, respectively. Many members of the nuclear receptor family form homo- or heterodimers, the DNA and the ligand binding domains are important in these processes.
Transcriptional activity of intracellular receptors can be up-regulated by phosphorylation of Ser residues in the N-terminal A/B domains by cyclin-dependent kinases, PKC, PKA, ERK, PKB/Akt, JNK/SAPK, p38-MAPK. AF-1 can be phosphorylated by CDK, ERK, JNK, p38-MAPK, PKB, while AF-2 by Src in case of ER. Down-regulation of transcriptional activity can be caused by phosphorylation of the DBD by PKC or PKA.

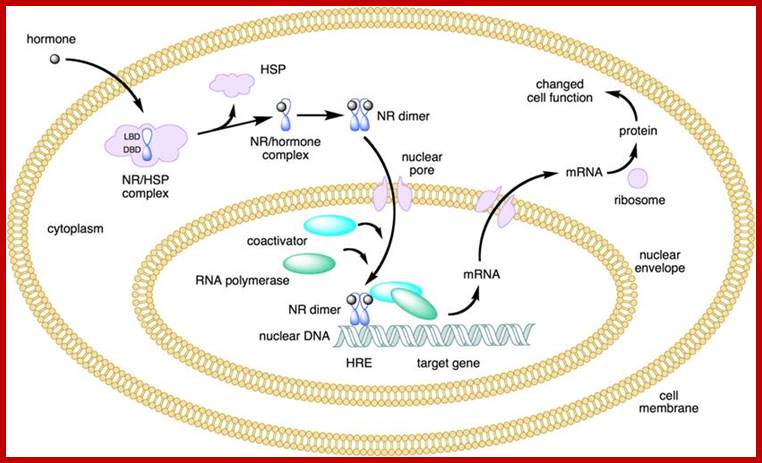
NR-Nuclear receptor; Some of the steroid hormones diffuse through lipid membranes and enter the cell interior, then the bind to steroid receptors and this complex enters nucleus and binds to specific DNA promoter sequences and activate gene expression; http://www.endocrinesurgeon.co.uk/ https://www.csblab.or.kr/trend
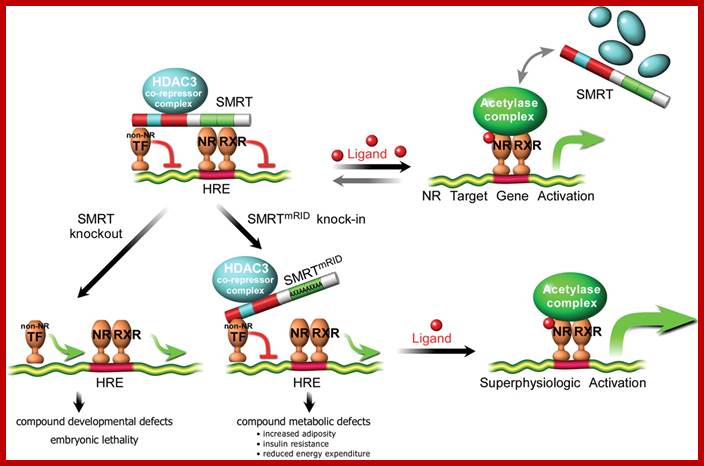
SMRT repression of nuclear receptors controls the adipogenic set point and metabolic homeostasis; a model for SMRT- mediated transcriptional repression.
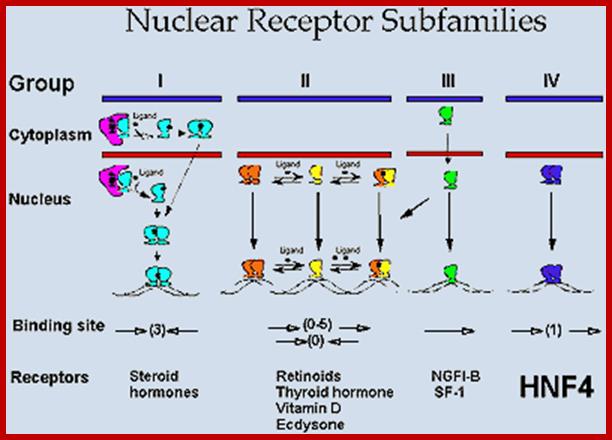
Nuclear Receptor Subfamilie;. Since HNF4 forms exclusively homodimers, is located only in the nucleus and binds DNA elements consisting of direct repeats, it defines a unique subfamily of nuclear receptors. This suggests that the mechanism of action of HNF4 is distinct from that of other receptors, which definitely appears to be the case. Adapted from Jiang et al. http://cbns.ucr.edu/
Intracellular Receptor families:
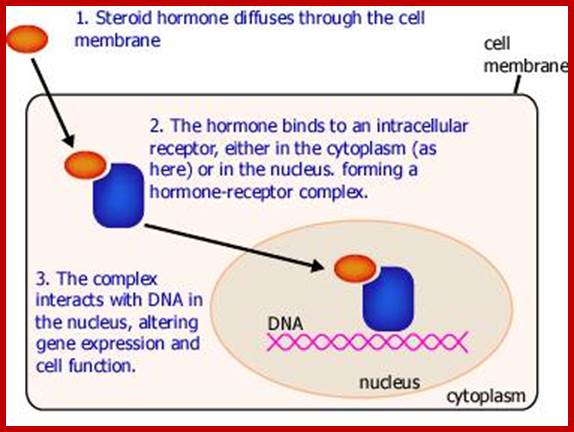
Some of the steroid hormones diffuse through lipid membranes and enter the cell interior, then the bind to steroid receptors and this complex enters nucleus and binds to specific DNA promoter sequences and activate gene expression; http://www.endocrinesurgeon.co.uk/
Cell Surface receptors:
Extra cellular signaling molecules have important roles in cellular development and function. For example platelet derived growth factor-a (PDGF-a) has an important role to play in the health of the cell. Similarly Tissue transforming growth factor-b (TGF-b has the ability to inhibit cellular proliferation of many types of cells including skin, epithelial and immune cells.
PDGF-b:
Signaling by the platelet-derived growth factor (PDGF) receptor; the unliganded receptor is monomeric and its tyrosine kinase catalytic activity is low (left). On binding to the receptor dimerizes; and its catalytic activity increases, and receptors Trans-phosphorylate each other on a number of different sites (specific), represented by pink circles (center). These phosphorylated sites (with one exception) serve to recruit cytosolic effector proteins (gray) that contain phosphotyrosine-specific modular binding domains (right). The exception is the activating phosphorylation, located on the catalytic domain of the receptor adjacent to the active site (red circle). Representative effectors depicted are: PI3K, regulatory subunit of phosphatidylinositol 3-kinase; GAP, Ras GAP, a GTPase-activating factor for Ras; PLC, phosphatidylinositol-specific phospholipase C-γ; Shp2, SH2-containing tyrosine phosphatase; Grb2, adaptor protein that recruits the Ras guanine-nucleotide exchange factor Sos. Src, Src-family non-receptor tyrosine kinases;

Action of PDFG and VEGF; http://www.ls.manchester.ac.uk/

PDFG signaling pathway; http://www.sinobiological.com/
The four PDGF ligands-PDGFA-D are inactive in their monomeric forms. The PDGFs bind to the protein tyrosine kinase receptors PDGFRA and PDGFRB. These two receptor isoforms dimerize upon binding the PDGF dimer, leading to three possible receptor combinations, namely -AA, -BB and -AB. a critical tyrosine residue, thereby "unlocking" the kinase, leading to full enzymatic activity directed toward other tyrosine residues in the receptor molecules as well as other substrates for the kinase.
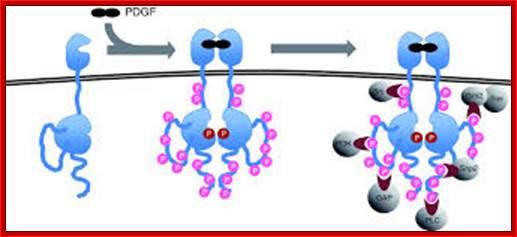
Dimerization can cause the activation of the kinase. Kinase activation is visualized as tyrosine phosphorylation of the receptor molecules, which occurs between the dimerized receptor molecules (trans phosphorylation). In conjunction with dimerization and kinase activation, the receptor molecules undergo conformational changes, which allow a basal kinase activity to phosphorylate http://jbiol.com/
When PDGF-b binds to its cell membrane receptor, it activates several components downstream, where phosphorylation by cellular domain of the receptor activates cytosolic s-mad proteins. Phosphorylated s-mad-2 interacts with phosphorylated s-mad-3 and produce dimers. The dimers in turn interact with unphosphorylated s-mad-4 factor and enter into the nucleus and activate several genes. PDFG receptor can also activated by Vascular Endothelial Factor (VGEF) acts using PDFG receptor.
1. Activates the expression of expression of p15; it is very important for p15 proteins block transition from G1 to S, so block the progression of cell cycle events at the earliest stage itself.
2. They also activate genes whose products are inhibitors of plasminogen activator activated protease. When the plasminogen-activated protease is active, it digests most of the extra cellular matrix, thus give way for the cancer cells to move from one place to the other. But the expression of an inhibitor prevents the digestion of ECM (extra cellular matrix proteins), so cell adhesion and cell-to-cell contact is maintained, that prevents cancer cell migration and expansion.
3. The TGF-b activated s-mad transcriptional factors also activate gene expression whose product reinforce extra cellular matrix, so no cancer cells can squeeze through the extra cellular space in the tissue and prevent the spreading.
4. On the contrary, if the extra cellular signal is absent or lost, none of the above said products are not produced and functions are not executed, thus cancer cells can easily migrate and spread through. It means this defect facilitates the spread of cancerous cells.
5. VEGF-A binds to PDFG receptors and activates PDGFG-AA to AB and BB receptors by activating tyrosine phosphorylation of PDFG-Rs.
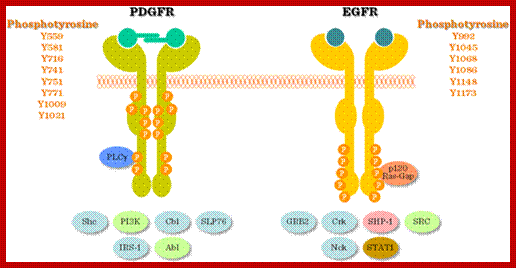
Growth factor receptors belong to the receptor tyrosine kinase family (for the details of growth factor receptor signalling -Receptor tyrosine kinases). http://www.tankonyvtar.hu/
Ligand binding leads to receptor dimerization, which induces phosphorylation of the cytosolic kinase domain and its activation (Figure above). Different receptors utilize different dimerization/activation strategies: for example PDGF is a dimer, which cross-links two cell surface PDGF receptor monomers; the binding of EGF to its receptor induces a conformational change, which promotes dimerization; FGF is complexed by heparin and cross links two FGF monomers; in case of insulin the receptor is already dimerized on the cell surface, ligand binding causes a conformational change and autophosphorylation (for more details on insulin signalling see next chapter).
Wnt pathway:
Wingless gene (Wnt) from Drosophila is called by different names in different organisms. Wnt. gene product is a secreted signaling molecule. It binds to cell surface Frizzled receptors that activate disheveled (Dvl) proteins in human beings; actually these are activated by casein kinases (Cs 1 and 2). This inhibits the activity multiprotein complex (consisting of b-Catenin, Axin, APC and glycogen synthase kinase (GSK-3b). Actually this complex when in normal situations phosphorylates b-Catenin and thereby it is subjected ubiquitination and proteosome-mediated degradation. It is important to note that the b-Catenin is involved with C. adherins in cell to cell adhesion.
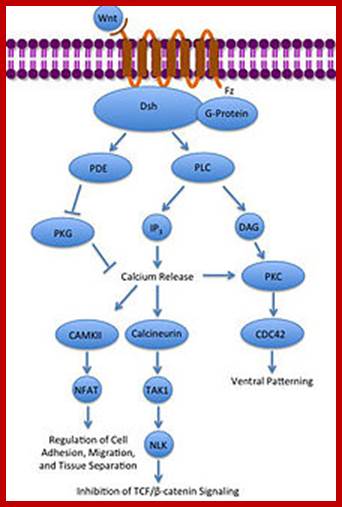
Noncanonical Wnt/calcium pathway; https://en.wikipedia.org
In the absence of Wnt signaling, the b-catenin accumulates in the cytoplasm and enters the nucleus and complexes with T-cell factors (TCF) and lymphoid enhancer factors (LEF) that results in the expression of cMyc and other genes required for continuous cell cycle events. The protein b-Catenin per se in association with other components is responsible for the activation of several genes, especially cMyc gene, responsible for inducing uninhibited cell cycle events.
The Wnt signaling pathways are critical in cell-cell signaling during normal development and embryogenesis and required for maintenance of adult tissue, therefore it is not difficult to understand why disruption in Wnt signaling pathways can promote human degenerative disease and cancer.
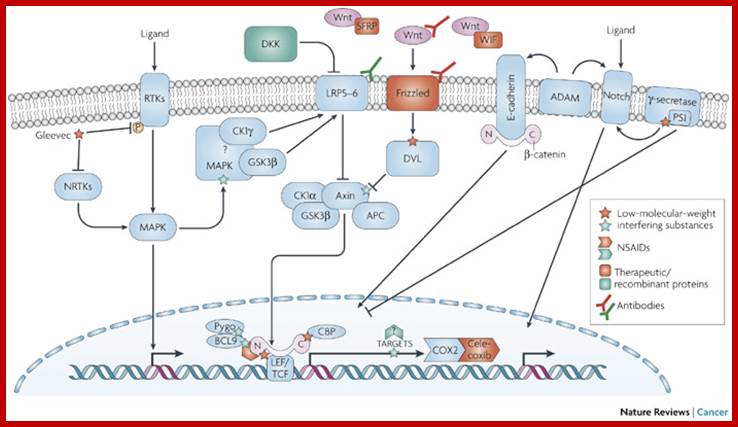
http://www.nature.com/nrc/journal
TGF-Beta Pathway:
Transforming growth factor (TGF) TGF-Beta regulates several processes in developing embryos, Including cell growth and differentiation, apoptosis and homeostasis. There are two types of receptors, Type I (seven types) and Type II. The receptors are known for their dual- specific kinases. Their cytoplasmic domain has week tyrosine kinase activity but strong serine/threonine kinase activity.
TGF-b canonical signal transduction pathway and transcriptional responses mediating TGF b growth inhibitory effects: At the cell surface, TGF-b assembles a complex of transmembrane receptor serine/threonine kinases (types I and II) and induces trans phosphorylation and activation of the type I receptor (T b R-I, ALK5) by the type II receptor kinase (T b R-II). Activated T b R-I phosphorylates the main TGF b downstream effectors, Smad2 and Smad3, at C-terminal serines. Activated Smad2 and Smad3 then associate with Smad4 and the complexes translocate into the nucleus and regulate transcription of target genes, involved in TGF b -induced growth inhibition. Down regulation of c-myc, CDC25A and Id family members and upregulation of p15 and p21 CDK inhibitors are key events in this response.
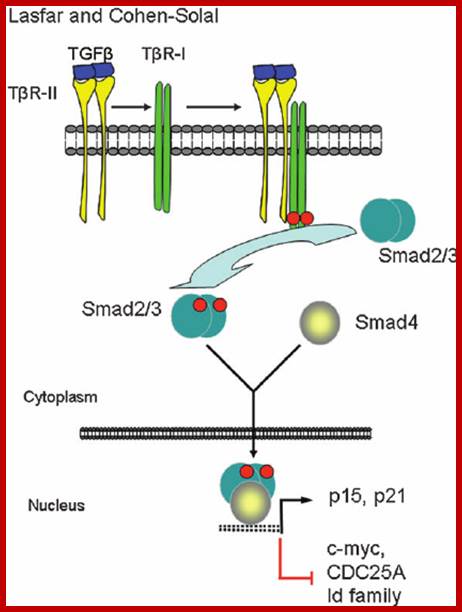
Transforming growth factor (TGF) b levels are elevated in the plasma of melanoma patients, especially those with metastatic lesions (1). In addition, TGF b 2 expression appears to be increased coincident with the development of invasive melanoma (2). https://www.researchgate.net/
Sffv (Spleen Focus Forming Viruses):
Spleen focus forming virus causes erythroleukemia. It is a retrovirus. Cellular erythroid precursors are activated by erythropoietin an hormone. The Epo binds to the receptors of Erythroid progenitor cells and activate gene-regulated transformation into fully developed erythrocytes. But when the virus infects, its capsid glycoprotein binds to the receptors of progenitor cells, where Epo binds. The binding of the capsid protein to the receptor is tight and it never releases from the receptor. This leads to the consistent signal induced activity; thus the cells without going through differentiation they multiply and expand their population, this actually causes erythroleukemia.

Signalling pathway activated by EpoR on erythrocytic cell in response to Epo. When Epo binds to erythrocytic progenitor cell, EpoR dimer undergoes conformational change and stimulates autophosphorylation of JAK2 kinase which inturn Plates tyrosine residues on EpoR intracellular domain. Stimulation of STATs5, AKT and ERK1/2 activates signaling cascade that leads to cellular differentiation and antiapoptotic effect and cellular proliferation. Effects of erythropoietin receptors and erythropoiesis-stimulating agents on disease progression in cancer
M Aapro, W Jelkmann, S N Constantinescu and B Leyland-Jones
http://www.nature.com/
HPV (Human Papilloma Virus):
Human papilloma virus on infection expresses its early gene product called E5 (44aa long). This protein has a transmembrane domain, thus it anchors in the membrane and dimerizes with another of its own, which then complexes with the cytosolic growth factors such as PDGF receptors on inner surface of the membrane. This causes sustained protein kinase activation of the receptor protein. This will have cascading effect leading to cell proliferation.
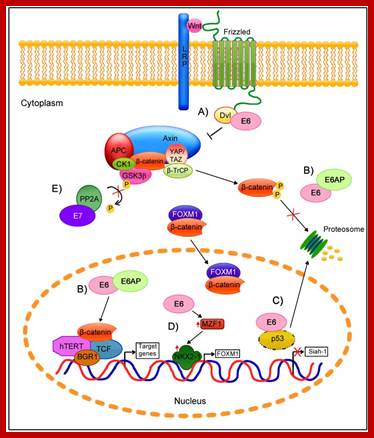
Regulation of the Wnt/β-Catenin Signaling Pathway by Human Papillomavirus E6 and E7 Oncoprotein; http://www.mdpi.com/
Signal Transducing Trans-membrane Receptor Proteins:
GPCR-Guanine protein coupled receptor, is seven fold transmembrane protein. The receptor is sensitive light, odors, pheromones, hormones and neurotransmitters. G proteins coupled receptors activate cAMP pathway and Phosphatidylinositol signal pathways. Research work on GPCR by Brain Kobilka and Robert Lefkowitz fetched Nobel prize in 2012. It is amazing that nearly 800 different human genes are predicted to code for GPCR. There are three subclasses, such as A-Rhodopsin like (largest),B-Secretin like and C- glutamate receptor like, others are Adhesion, Frizzled/Taste2 type and secretin like and few unclassified. Class A (or 1) (Rhodopsin-like), Class B (or 2) (Secretin receptor family), Class C (or 3) (Metabotropic glutamate/pheromone), Class D (or 4) (Fungal mating pheromone receptors), Class E (or 5) (Cyclic AMP receptors) and Class F (or 6) (Frizzled/Smoothened).
The protein has extracellular N- terminus followed by seven TM (7-TM) alpha helixes connected by three extracellular and three intracellular loops and its C- terminus (KKKRRK domain) is cytoplasmic end. It has cavity like structure to which ligands bind

https://en.wikipedia.org
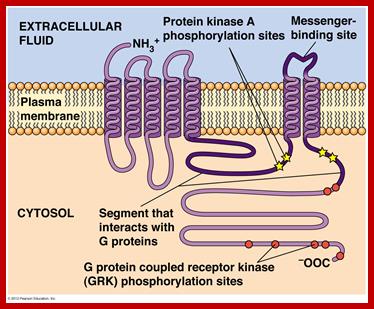
Signal transduction Pathway-GPCR; http://www.mun.ca/biology
There are more than 35-40 different ligands. They bind and activate this GPCR receptor. GPCR protein is bound to a trimer of alpha, beta and gamma subunits. It has Phosphatidylinositol and cAMP pathways. The alpha and gamma subunits are bound to PM- by lipid anchoring.

Activation of the G alpha subunit of a G-protein-coupled receptor
In unstimulated cells, the state of G alpha (orange circles) is defined by its interaction with GDP, G beta-gamma (purple circles), and a G-protein-coupled receptor (GPCR; light green loops). Upon receptor stimulation by a ligand called an agonist, the state of the receptor changes. G alpha dissociates from the receptor and G beta-gamma, and GTP is exchanged for the bound GDP, which leads to G alpha activation. G alpha then goes on to activate other molecules in the cell.
© 2002 Nature Publishing Group Li, J. et al. The Molecule Pages database. Nature 420, 716-717
(2002). ![]()
G-protein’s alpha subunit binds to either GTP or GDP, when active it binds to GTP and when inactive it binds to GDP; in active form it is bound to GTP.
Activation of a single G protein activates the production of more than hundreds of second messenger molecules; the second messengers such a cAMP, Diacylglycerol (DAG) and Inositol (IP3), when activated initiate coordinated intercellular signaling pathways. One important, but common target is adenylyl cyclase a membrane associated enzyme. The cAMP has multiple functions on target substrate.

The relationships of G proteins to the plasma membrane:
In this diagram of G-protein-coupled receptor activation, the alpha, beta, and gamma subunits are shown with distinct relationships to the plasma membrane. After exchange of GDP with GTP on the alpha subunit, both the alpha subunit and the beta-gamma complex may interact with other molecules to promote signaling cascades. Note that both the alpha subunit and the beta-gamma complex remain tethered to the plasma membrane while they are activated. These activated subunits can act on ion channels in the cell membrane, as well as cellular enzymes and second messenger molecules that travel around the cell.
Receptor Kinases:
Many growth hormone receptors are transmembrane proteins with kinase activity at their cytosolic ends, but this need not be the case in all other membrane protein receptors. Epidermal growth factors, Platelet derived growth factors, Colony stimulating factors and many such factors act as mitogens. They bind to their membrane receptors and activate specific kinase activity.
EGF-Receptor Tyrosine Kinase:
Epidermal growth factor (EGF) receptor is a class-I receptor having N-terminal extracelluler domain and a cytoplsmic domain with Tyrosine Kinase activity. This gene is also called c-erb-B. It has single trans-membrane domain. Ligand binding leads to dimerization and activation of Kinase activity at cytoplsmic side. This can lead to autophosphorylation at it cytosolic domain. The cytosolic domains may contain protein-protein interacting domains such as SH2 and SH3. The active cytoplsmic domain transduces the signal to other proteins in the form of phosphorylation of target proteins at tyrosine moiety. There are 18-20 receptor kinases. The ligands are EGF, Insulin, PDGF, VEGF, FGF, HGF, Trk, Eph, AXL, LTK, TE, ROR, DDR,RET, KLG, RYK and MuSK.
But oncogenic proteins have deletions, either in the form of missing external domain or internal cytosolic domain or both, yet transmembrane domains remain as dimer and the protein remains active even without ligand binding.
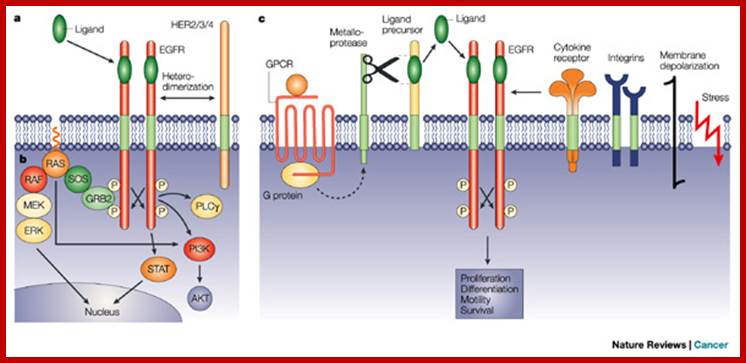
Ligand binding to the receptors induces
for dimer formation; this leads to cross –autophosphorylation of tyrosine in
cytoplasmic domains which function as docking sites for downstream signal
transduction. This leads to signaling cascade RTK–GRB2–SOS–RAS–RAF–MEK–ERK, PI3K–AKT,
PLC![]() and STAT pathways.
and STAT pathways.
Loss of external domain makes cytosolic domain dimerizes and active with out the ligand. Similarly loss of cytoplasmic domain makes the transmembrane domain dimeric and active without the binding of ligand. This provides tremendous mitogenic stimulus for the cell enter into cell division mode. The mutated gene is called c-oncogene or if the virus carries it, then it is called V-erbB1. Another epidermal growth factor gene is c-erbB2, it is a related to erbB1. A mutation in the transmembrane domain renders it as a dimer and to be active all the time.
Colony stimulating factor-1: Receptor tyrosine kinase:
This is another receptor protein tyrosine kinase. In the case of colony stimulating factor receptor (csf1), coded for by C-fms gene, mediates the action of colony stimulating factor. This is a macrophage growth factor that stimulates growth and maturation of myeloid precursor cells into myeloma. Mutation in the extra cellular domain renders it to be dimeric and oncogenic. This kinase protein remains active all the time, even with out the ligands. Oncogenicicty is enhanced by mutations at cytoplsmic domain for the cytoplsmic domain has inhibitory activity. As the proto-oncogene is involved in development of a particular type of cell populations, its mutation leads to unrestricted growth and proliferation. Mutation in the Csf-1 gene makes the protein ever active and the kinase activity has cascading effect on cellular components.
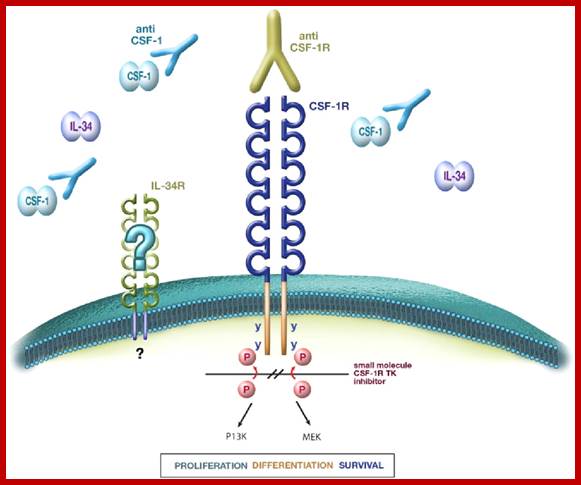
Therapeutic applications of macrophage colony-stimulating factor-1 (CSF-1) and antagonists of CSF-1 receptor (CSF-1R) signaling; www.blood journal.org

Schematic representation of
the receptors for macrophage colony-stimulating factor (M-CSF), granulocyte/macrophage CSF (GM-CSF) and
granulocyte CSF (G-CSF), showing subunits and some structural elements. The M-CSF receptor (CSF1R; also
known as FMS) is a homodimeric type III receptor tyrosine kinase; the GM-CSF
receptor (CSF2R) consists of a unique ![]() -chain and a common
-chain and a common ![]() -chain (
-chain (![]() c), through which
signalling occurs; and the structure of the G-CSF receptor (CSF3R) is typical
of the type I cytokine receptor family (adapted from Refs 18,21). The
intracellular signalling pathways that can interact with the cytoplasmic
regions are not shown (see main
text). FBN III domain, fibronectin type III domain;
http://www.nature.com/
c), through which
signalling occurs; and the structure of the G-CSF receptor (CSF3R) is typical
of the type I cytokine receptor family (adapted from Refs 18,21). The
intracellular signalling pathways that can interact with the cytoplasmic
regions are not shown (see main
text). FBN III domain, fibronectin type III domain;
http://www.nature.com/
Trk gene:
Human carcinoma in many cases has been traced to mutation in Trk gene. The gene product is Receptor tyrosine kinase protein. Mutation due to abnormal chromosomal translocation, the extra cellular domain of the protein is replaced with the N-terminal part of non-muscle Tropomyosin. The Tropomyosin domain forms coiled coils at the extracelluler domain and thus lead to dimerization at its cytosolic side. In normal situation the dimer receptor is activated by the nerve growth factors (neurotrophins), and the activation leads to dimerization at cytosolic level and activation of the receptors RTK. In this mutation the receptor is constitutively activated with out the stimulus by NGH.
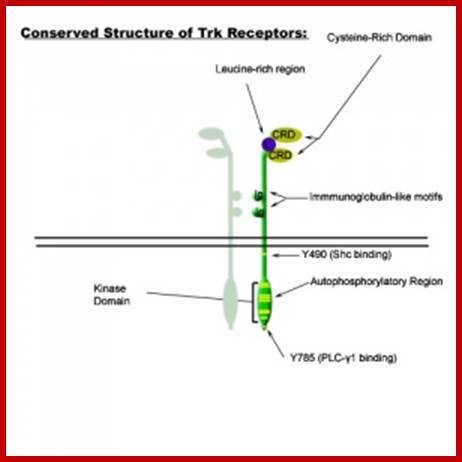
Common structural features of Trk receptors: a Trk in dimer complex with various conserved regions and residues. Each Trk (A, B or C) is composed of a number of highly conserved elements. Variability mainly occurs in the ligand binding regions. Variations in binding site affinity can affect phosphorylative domain regions and adaptor protein association differently, thus stimulating different signalling events; Specificity of Trk receptors for neurotrophin ligands: NGF binds significantly with TrkA. BDNF & NT-4 bind most significantly with TrkB. NT-3 binds most signifcatly with TrkC, however, NT-3 also elicits low-affinity binding with Trk's B and A.
;https://cellbiology.med.unsw.edu.au
Similarly mutation in Glial Neurotrophic receptor kinase, by valine/glutamine, makes the external domain dimeric and active, such mutations can cause endocrine neoplasia. Mutation that substitutes Valine to glutamine in Human epidermal receptor kinase Her2 makes it an oncoprotein called Neu. A mutation causing substitution from Valine to glutamine makes this protein ever active. Even under normal conditions over expression of this protein renders it as one of the oncogenic factor responsible for causing human breast cancer. (Note that a monoclonal humanized antibody against Her2 receptor makes the receptor internalized selectively and kills the cancer cells).

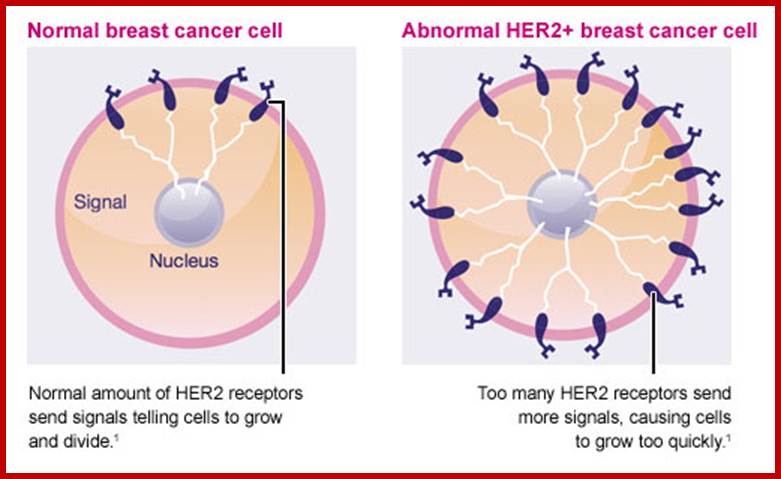
http://www.aboutcancer.com/
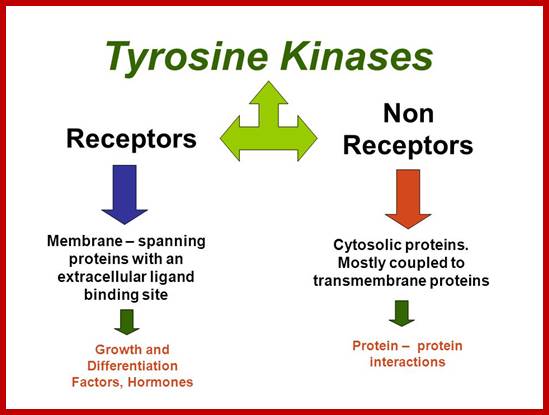
Non-Receptor Tyrosine Kinase:
Ras Gene:
It is one of the important non-receptor kinase gene mediates between receptor and cytosolic kinases. It connects the plasma membrane receptor to cytoplasmic transducers or targets. Ras is a family of genes, like H-Ras carried as a V-oncogene by Harvey murine Src virus, H-Ras, K-Ras is carried as V-Ras by Kirsten murine Src virus; they are closely related genes whose products are of 21 KD proteins.
http://slideplayer.com.slide
It is a cytosolic G-protein, but it is activated by transmembrane receptor tyrosine protein kinase. It connects the plasma membrane receptor to cytoplsmic transducers or targets.
http://www.cytoskeleton.com/
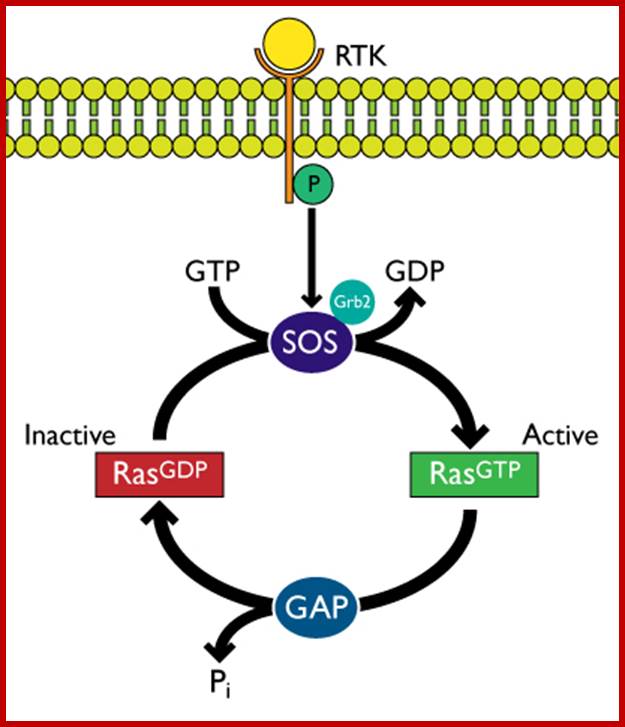
Receptor Tyrosine kinase-K-RAS signaling; http://www.cytoskeleton.com/
http://www.personal.psu.edu/
Normal Ras gene product is a monomeric GTP biding-protein involved in signal transduction from receptor tyrosine kinases to down stream cytosolic kinases. Ras by itself is a tyrosine protein kinase. Though this protein is cytosolic, it is anchored to cytoplasm through Farnesyl, an isoprene. Farnesyl transferase is responsible for the addition of Farnesyl to Ras. An inhibitor to the addition of farnesyl group prevents this. Use of such inhibitors, on Ras transformed cultures, shows reduction in abnormal proliferation cells.
The Ras is activated by receptor tyrosine kinases through adaptor proteins such as Grb2 and Sos. When Ras is bound to GTP, it is active, after hydrolysis of GTP to GDP, facilitated by GAP; the enzyme remains inactive till it is replaced with another GTP through GTP exchange factor (GEF). It has intrinsic GTPase activity. GAP stimulates Ras’s GTPase activity. In its active state this protein is involved in Ras-MAP kinase pathway.
When this gene is mutated to RasD , i.e. substitution of amino acid glycine at 12 or glutamine at 61 with any other amino acid, except proline, it remains active, for the bound GTP remains bound for a long time, for the bound GTP is cleaved very slowly thus the duration of signal transduction will be longer than required. This protein remains active even in the absence of stimulation by growth promoting hormones.
Onco-Ras containing DNA can easily transform primary cells into oncogenic cells. Just over expression of the c-oncogene by 20% more than the normal, can induce cancer. The over expression or excess expression can be due to duplication of genes by translocation or the gene placed under promoter or enhancer of another gene, where the Ras genes is expressed more than the normal level of expression and it causes cell transformation into tumors. Ras onco-protien is expressed in many tumor tissues such as bladder, colon, and mammary and skin cancers.
1. One MAP kinase phosphorylates Transcriptional factors (TFs) and induces expression of important genes required for cell division and differentiation steps. Mutation in the Ras short-circuits the first part of the pathway, because the mutated Ras remains active all the time and causes over expression of genes.
2. Another pathway is mutation in GTPase activating enzyme (GAP), so the loss of GAP leads to sustained Ras activation and it leads to down stream activation pathway. Individuals with Neurofibromatosis have inherited single mutant of NF1 allele. Further mutations leads to the development of benign NF tumors in sheath cells that surround Nerves. Receptor tyrosine kinase (RTK) pathway of signal transduction starts from the mitogenic ligand binding to the receptor. The activated receptor then activates Ras through adaptor proteins such as Grb2 and Sos, which in turn activate Ras. The signal transduction pathway continues from Ras to Raf to Mek to Mapk and MaPkk and so on, ultimately nuclear factors, one such factor id NF1 and its family of proteins. When the are activated they enter into the nucleus and activate hundreds of genes among them some are cell cycle progression protein genes.
Mitogenic factors induced RTK-Ras-MAP Kinase Pathway:
Extracellular signal-regulated kinases (ERKs)- The first mitogen-activated protein kinase to be discovered was ERK1 (MAPK3) in mammals. EGF and PDFG and many act as mitogens.

Mitogens trigger cell divison; http://tbl.med.yale.edu


http://tbl.med.yale.edu/ Mitogens trigger cell division
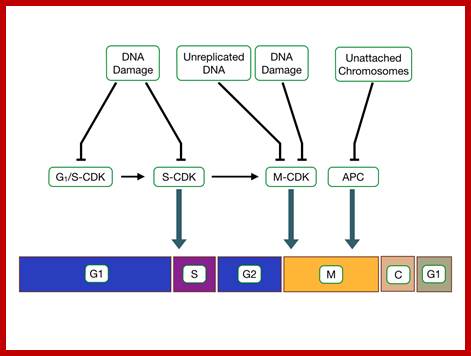
Cell cycle check points;, the said proteins prevent not needed cell division/factors http://tbl.med.yale.edu/;
Mitogens activate as they bind to cell surface receptors containing MAPkinase. MAPkinase is a powerful kinase that activates many cellular functions such as cell proliferation, gene expression, differentiation, mitosis, cell survival and apoptosis. There are 16 or more MAPkinases.
The cascading effects of many mitogenic signaling molecules, first activate the membrane receptors, most of them have kinase activities. If we take RTKs the activated receptor kinases activate Ras through adaptor proteins such as Grb2, Sos and then to Ras. The Ras has down stream activation of Raf, then Mek and Mapk and MeKK thus leads to the activation of several nuclear transcriptional factors. It is that leads to the activation of several genes especially related to the cell cycle events. When such genes are continuously expressed, the cell cycle events progress with out any check or control; it is that leads to cell transformation into a cancerous cell. In this cascade of related steps, if any one of the proteins is rendered constitutively active, the cell gets transformed.
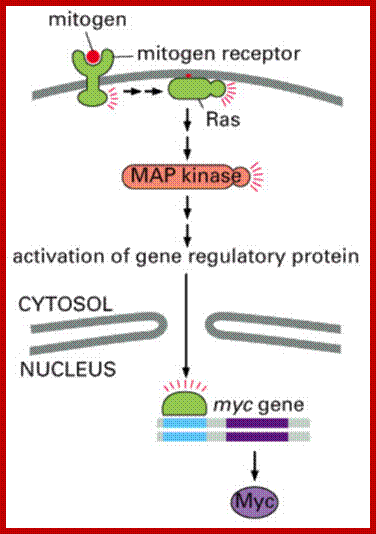
Cell Cycle MAP Kinase; https://quizlet.com
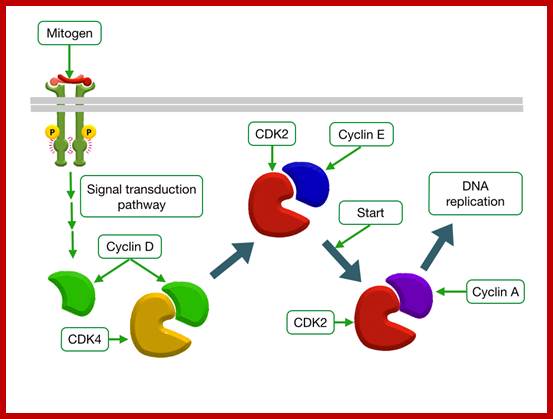
Mitogen triggered Cell divison; http://tbl.med.yale.edu;
 Epidermal
Growth Factor Increases Transcription of CyclinD; http://tbl.med.yale.edu/
Epidermal
Growth Factor Increases Transcription of CyclinD; http://tbl.med.yale.edu/
V-Src:
A mouse retroviral gene encodes constitutively active Raf, a serine/Threonine kinase, which acts in between Ras and MAP kinase. Another is erk oncogene; in avian sarcoma virus the gene product contains SH2 and SH 3 domains, which is similar to GRB2 an adaptor protein that also functions between RTK and Ras. The SH2 and SH3 domains mediate specific protein aggregation that mediates signaling for cellular events. Over expression of erk leads to over aggregation and inappropriate signaling for the growth; this leads to metastasis abilities of cancer cells. Now it is known that there are specific genes for cancerous cells leading to metastasis. Ras is the most potent oncogenic protein. If certain oncogenes are over expressed in transgenic mice, they can cause cancer. For example if transgenic mice with Myc alone, few out of 100 mice get at the end of 100 days, but if Ras is expressed in transgenic mice, at least 40-50 percent of the mice get cancer in hundred days. If both the genes are over expressed almost all mice get cancer. This illustrates the potentiality of the Ras gene as the most potent cancer gene, provided it is made to express in abnormal amounts. This is because the Ras gene has the ability to express the pathway of cyclin D1 synthesis as the Myc gene does.
RTK > grb2 > Sos > Ras > Raf > Mek. Mapk > Mapk >>> NFs
This is a modified cellular gene (c-src), but found in virus Rous sarcoma, discovered by Peyton Rous in 1911. The Src is not a GTP binding protein. The V-Src gene carried by the virus is a truncated gene where the sequences for 19 amino acids at the carboxyl end are deleted. This viral infection causes cancer called sarcoma. The counter part of this gene in cells is proto or C-Src. The C-Src gene product has the ability, in its active state, to phosphorylate certain target proteins (motif dependent manner), so the protein is called Tyrosine Protein Kinase (TPK).
In viral infected cells, the gene is over expressed so it causes over-phosphorylation of some proteins involved in cell cycle events. Phosphorylation and dephosphorylation events can lead to a cascade of signal transduction pathway.
The normal cellular Src protein, between 80 and 516 amino acids, has common sequence with other but related proteins such as yes, fgr, fps/fes, abl and ros. These are also cytosolic kinases. But Src protein is anchored on to the inner surface of plasma membrane at N-terminal region via myristoylation (14 C). From 80 to 250 it has modulator domains called SH2 and SH3 (SH means Src- domains, because these were first observed in src protein so the names). The SH2 and SH3 domains are protein –protein interacting domains and they are important. The modulator regions play important role in oncogenicity. Mutation in SH2 region reduces oncogenicicty and mutation in SH3 increases oncogenicity.

http://www.mdpi.com/
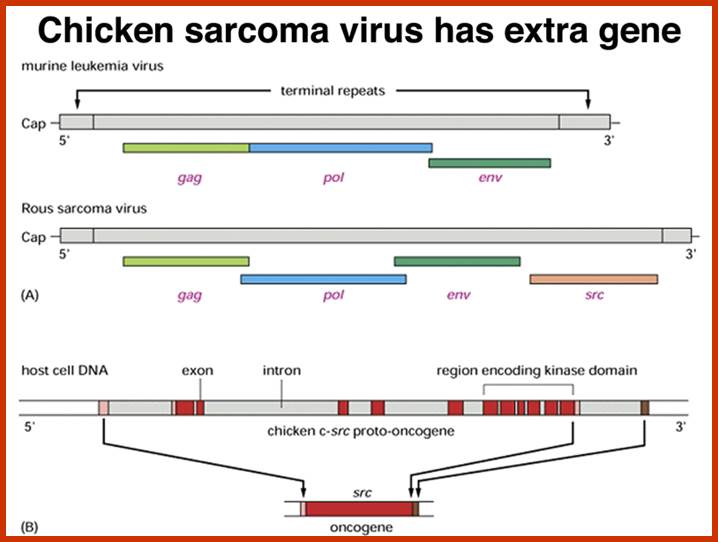
Compared to closely related retroviruses like murine leukemia virus, there is an extra gene in Rous sarcoma virus, named src. The src gene carried by Rous sarcoma virus causes tumors; if it is inactivated, the virus does not induce sarcomas. Surprisingly, the src gene carried by the virus (v-src) is a mutated version of a gene found in normal cells (c-src). The protein encoded by the src gene is a tyrosine kinase. The v-srcgene has a gain-of-function mutation that makes the kinase constitutively activated, not subject to the regulation that controls the c-src gene.; http://www.discoveryandinnovation.com/BIOL202/notes
The normal C-Src protein kinase contains one tyrosine at 416 and another at 527. The catalytic domain lies between 250 to 516, and the suppressor domain at 616 to the carboxyl end. Phosphorylation of Src at 416 makes it active, but phosphorylation at 527 represses its kinase activity. When the protein is phosphorylated at 527, the Tyrosine at 416 is tucked inwards and not accessible for phosphorylation. Mutation at tyrosine at 416 makes it inactive, but mutation at Tyrosine 527 to phenylalanine or the deletion of 17-19 amino acids at carboxyl-end of the protein makes the Src protein ever active.
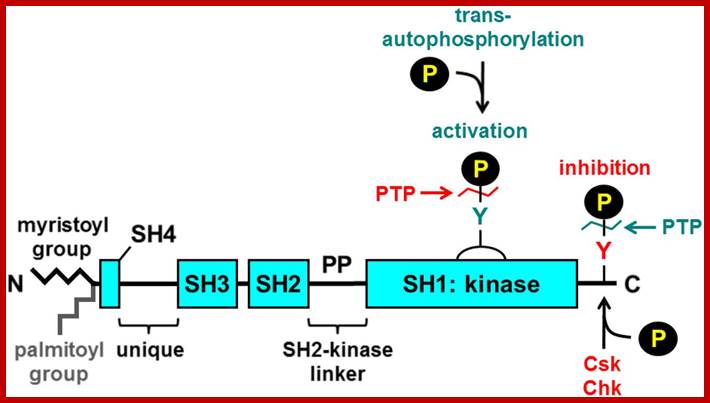
http://www.bloodjournal.org/
When tyrosine at 527 is phosphorylated, this end interacts with its own SH2 domain. It masks the 416-kinase domain. When the transmembrane receptor is activated, its cytosolic domain gets autophosphorylated; this transforms the protein in such a way the C-end of the receptor protein interacts with SH2 domain of C-Src, and releases the C-end of the Src free, which leads to unmasking of activation domain and the tyrosine at 416 gets phosphorylated, thus the C-Src becomes active.
In V-Src or mutated cellular Src, the last 17-19 amino acids are missing, so the 416 amino acid is free for phosphorylation and the Src becomes active and remains active with out the activation of its receptor kinase protein. This has down stream effect on cellular components. The Src protein is associated with cytoplsmic cytoskeletal structures; this has morphogenetic effect. In few other cases where the cell that is infected with S40 or its related DNA viruses, the T-antigen produced by the viral genome binds to the C-terminal region of the Src; this makes the normal Src to be active all the time. When the Src is continuously active it phosphorylates many of its targets and the cell gets transformed for the cell cycle events get deranged to proliferation without control.
Over expression of the unmodified C-Src protein can also be achieved by retroviruses. When such viruses carrying the intact gene is integrated into host chromosome and if the Src gene is under the control of viral LTR sequences, the src protein is all the time is transcribed. A similar over expression of the src can also be achieved by the translocation of the chromosomal segment containing Src gene to a site next to an activator or enhancer sequence of another gene. Again this effects continuous expression of the Src protein. Any expression more than 10% above the normal triggers the transformation.
Chromosomal aberration:
Chromosomal aberrations in terms of Deletions, Duplications, Inversions and Translocations can change the positions of certain genes and such positional effect when an cancer gene is place downstream of an enhancer domain can cause cancer. It is interesting to note a very significant number of cancers are due to chromosomal non-reciprocal translocation.
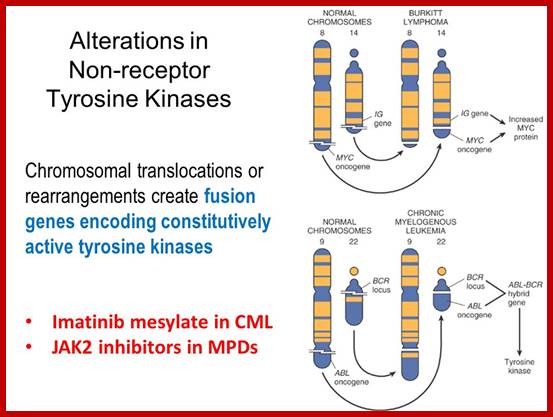
Myelogenous Leukemia:
Translocation abnormality-1:
This is a classic example of cancer due to illegitimate translocation, which creates a hybrid gene containing coding region for the kinase from the C-abl gene and a promoter region from Bcr gene. This is also a good example where c-genes by abnormal translocation and integration at different sites can make genes, especially of cell cycle related ones into ever active. One such gene combination is of C-abl and Bcr (break point cluster gene) where N-end of Bcr fuses with COO terminal region of the C-abl gene to produce hybrid gene.
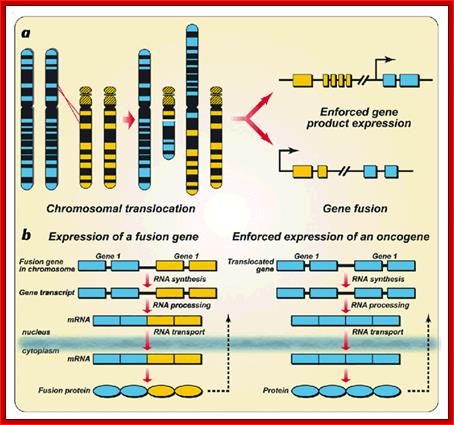
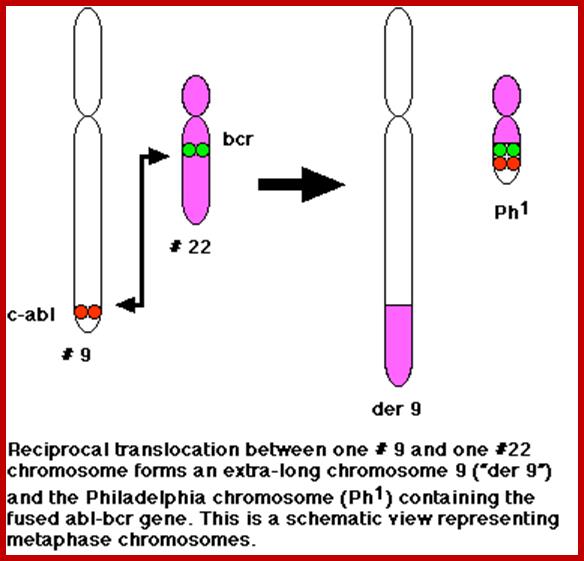
Myelogenous Leukemia (CML); http://users.rcn.com/
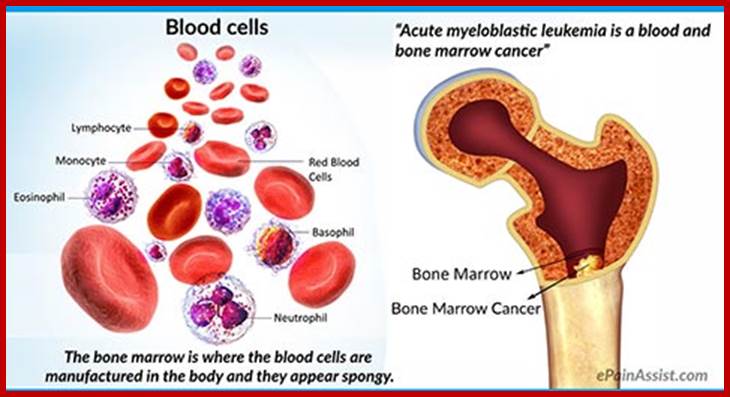 Acute myeloid leukemia is another name for
acute Myeloblastic leukemia. It is also called acute nonlymphocytic leukemia
and acute granulocytic leukemia;
Acute myeloid leukemia is another name for
acute Myeloblastic leukemia. It is also called acute nonlymphocytic leukemia
and acute granulocytic leukemia;
http://www.epainassist.com/
A 550-kbp long region of chromosome-9 carrying C-abl is transferred next to Bcr in the chromosome 22; a part of the Bcr is translocated to chromosome 9. The Bcr region is about 5.8Kbp within which C-abl gets integrated. The fused gene product is about the size of 210KD (70Kd is Bcr and 140 Kd is C-abl). The N-terminal part of the Bcr produces coiled-coils. Part of the C-Abl encodes with protein tyrosine kinase activity. The chimeric protein associates into a tetramer, which has constitutive Abl-Kinase activity. This onco-protein acts as cytosolic kinase. Through SH2 and SH3 domains Bcr binds to many intracellular signal proteins and phosphorylate them.
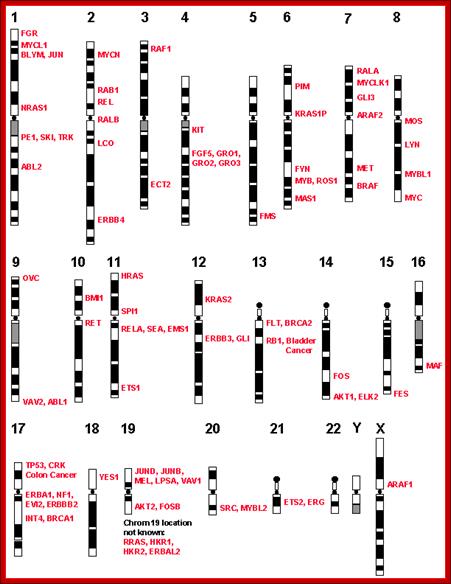
Few cancer genes’ loci on human chromosome; http://www.suggest-keywords.com/
Due to such kinase activity of proteins such as Jak2 protein tyrosine kinase, Stat-5 (transcription factor) and PI-3 kinase (Tri-Phospho Inositol Kinase) other downstream proteins become active even in the absence of growth factors or mitogens. This leads to activation of many genes related to cell cycle through phosphorylation of specific nuclear factors and the activation of Cdk-cyclin kinases systems.
A chromosomal translocation that produces Bcr-Abl combination in Hematopoietic cells is a diagnostic tool called Philadelphia chromosome. This results in chronic human Mylogenous leukemia (CML). In these patients differentiated Granulocytes (a type of WBC cells) expand in numbers. A second mutation in the same cell leads acute leukemia, which kills the patient.
Burkit’s Lymphoma- A Translocation abnormality-2:
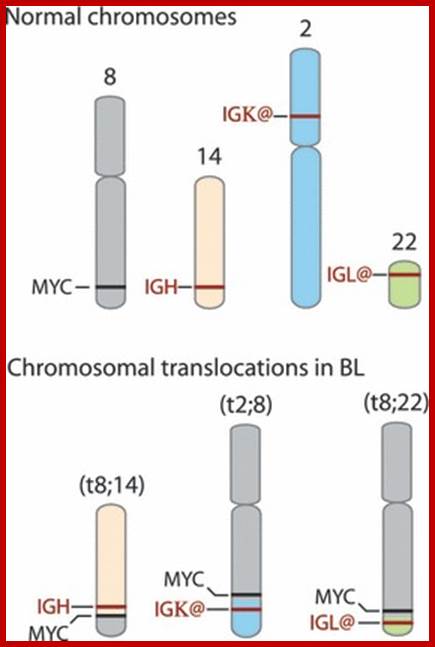
C-Myc gene is located on chromosome 8 and IgG-V/H locus is located on chromosome 14. In somatic cells some times nonreciprocal translocation between these two chromosomes result in the exchange of chromosomal segments resulting in positioning of cMyc next to immunoglobulin heavy chain enhancer region in the 14th chromosome.

Up to 70% of all human malignancies show elevated
expression of MYC. MYC is a pleiotropic transcription factor involved in many
aspects of cellular development and physiology. Besides direct regulation of
target genes involved in proliferation and growth MYC is implicated in
controlling the complex networks of microRNAs and apoptosis mediators. The mode
of MYC deregulation varies between different tumor entities. In most types of
cancer high MYC levels are secondary to alterations in cell signaling pathways,
leading to enhanced proliferation of the transformed cells. In some hematological
malignancies, like Burkitt lymphoma (BL) and subsets of diffuse large B-cell
lymphomas, elevated MYC levels are a direct consequence of genomic aberrations
involving the MYC locus. BL is considered the prime example for MYC-induced lymphoma
genesis. In comparison to other hematological malignancies it has the highest
MYC-expression and is often connected to Epstein-Barr virus (EBV) infection.
Over the past five decades BL has provided an invaluable tool for the entire
discipline of oncology, helping to decipher many aspects of tumor biology. This
review summarizes recent advances in the research on MYC-induced lymphoma
genesis, focusing on the regulation of microRNAs and apoptosis, and possible
contributions of EBV for lymphoma development.
Advances in understanding of MYC-induced lymphoma-genesis; https://www.researchgate.net
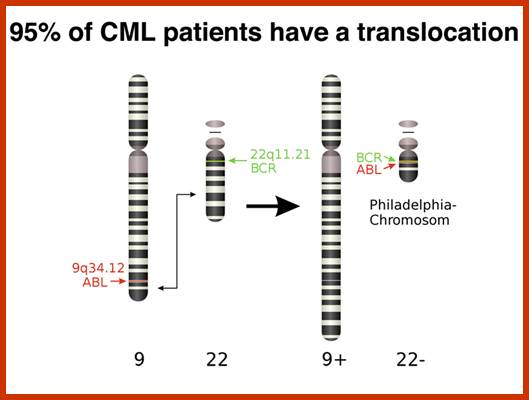
There is no change in ploidy associated with a reciprocal translocation, so there must be something oncogenic about the rearrangement breakpoint. This is shown below. All occurrences of the Philadelphia chromosome in CML have breakpoints somewhere in a gene called bcr1(for breakpoint cluster 1) that creates a gene fusion with the ABL gene. The ABL gene is a receptor tyrosine kinase. The fusion gene expresses a constitutively-activated receptor tyrosine kinase like the v-erbB oncogene. http://www.discoveryandinnovation.com/
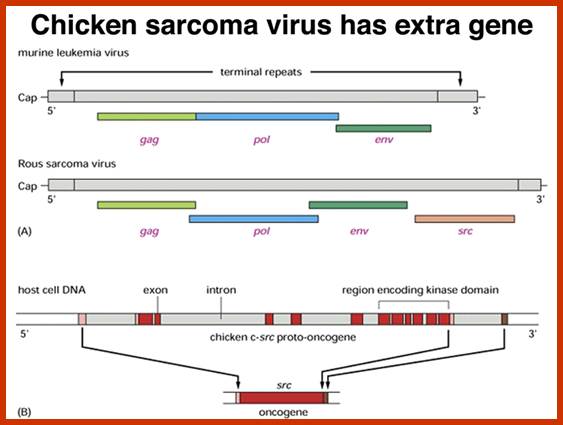
Compared to closely related retroviruses like murine leukemia virus, there is an extra gene in Rous sarcoma virus, named src. The src gene carried by Rous sarcoma virus causes tumors; if it is inactivated, the virus does not induce sarcomas. Surprisingly, the src gene carried by the virus (v-src) is a mutated version of a gene found in normal cells (c-src). The protein encoded by the src gene is a tyrosine kinase. The v-srcgene has a gain-of-function mutation that makes the kinase constitutively activated, not subject to the regulation that controls the c-src gene. http://www.discoveryandinnovation.com/
Thus cMyc is constitutively expressed. cMyc is known to induce the expression of cyclin-D and related genes, which are essential for the progression of cell cycle from G1 to S phase; thus induce cell transformation into cancerous cells.
B-Cell Lymphoma-translocation abnormality-3:
IgG-H chain enhancer region is located in chromosome 14 and Bcl2 gene on chromosome 18. Abnormal translocation results in exchanging of the genes resulting in the transfer of Bcl2 next to the Enhancer region of IgG-H chain on chromosome 14, this leads to consistent expression of Bcl2, which causes cancer.
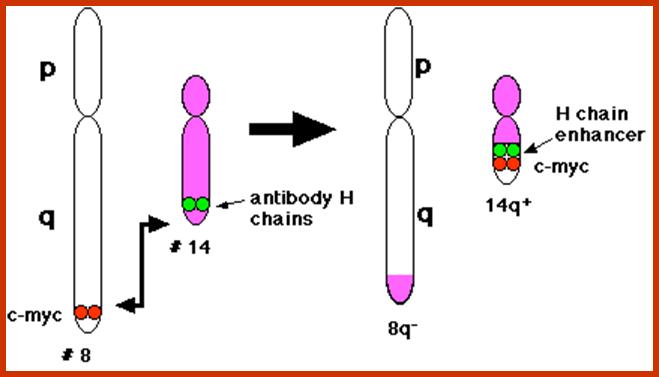
Burkitt's lymphoma, a reciprocal translocation (designated t(8;14) has moved the proto-oncogene c-myc from its normal position on chromosome 8 to a location close to the enhancers of the antibody heavy chain genes on chromosome 14.; http://www.biology-pages.info/B/BurkittLymphoma.html
Gene amplification as a cause for Cancer:
Due to unequal translocations certain genes are duplicated or become multiple copies. For example in breast cancers Her2 is found in multiple copies, this results in over expression. Similarly some genes at the vicinity of DHFr loci get amplified during replication, if the gene next to it happens to be a protein kinase gene, then this leads to transformation of Cells. In fact in breast cancer cells, it has been found 40-50 genes exist in multiple copies and five or more genes are found over express.
Micro array Assay:
Search into the analysis of genes expressed in cancer tissues, and how they differ from the normal tissues, DNA microchips were used and hybridized with labeled cDNA from the normal and malignant cancer tissue, it has been found fifty or more genes are found to be over expressed, which are not so in control tissues.
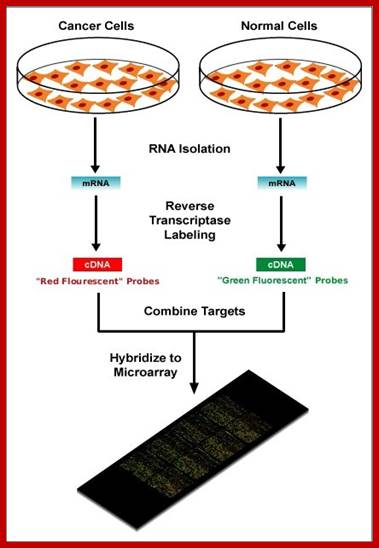

https://elodiebrans.wordpress.com/
Mutations among proto-oncogenes; https://oncogenesandcancer.wordpress.com
But surprising outcome of this experiment is hundreds of genes expressed in control tissues are found to be repressed in cancer cells. It means, cancer is not just due to the expression of cancer specific genes, over expression of some but suppression of a whole lot of other genes. A very important out come of these studies is different types of cancers emanating from different tissue types, have the same kind of genes. The names for these genes have been attributed for the discovery of a gene in specific cancer type for the first time, is the reason for nomenclature. If one makes a survey of different types of cancers and the genes responsible for, one finds genes expressed in most of the cancer types are found to be common. One important out come of these studies is that the genes which control cell cycle check points have lost their functions and another set of genes that have lost their function is tumor suppressor genes.
APC Genes:
Adenomatous Polyposis Coli cancer is due to mutation in both allelic APC genes, was found to be responsible for colon cancers leading to the development of polyps and also in developing benign Adenoma and Carcinomas. Polyps have high potentiality to generate cancerous tissue. APC is one of the tumor suppressor genes, which inhibits progression of certain types of cells through cell cycle and proliferation.
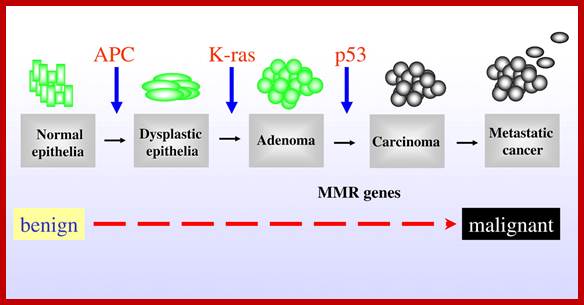
Mutations in APC, Kirsten-ras, and p53—alternative genetic pathways to colorectal cancer; The proposed order of mutations in APC, K-ras,p%# and DNAMMR genes is illustrated above the development of Cancer. http://www.pnas.org/
APC inhibits Wnt protein (Wilmer tumor). The Wnt protein has the ability to in a series of reactions phosphorylate b-catenin for ubiquitn mediated degradation. In the absence of APC gene b-catenin enters the nucleus and in association with other factors such as leaf activates several gene expressions. Among them Myc gene is one. Note b-catenin is also the protein involved in cell-cell adhesion. The Myc is a transcription factor. Myc is responsible for the expression of many genes involved in the progression of cell cycle especially transition from G1 to S phase. APC inhibits Wnt protein activity thus Myc is not produced and no activation of genes involved in the progression of cell cycle from G1 to S; thus APC gene is tumor suppressor.
APC is relatively a large gene codes for a transcript of 9000 ntds long, contains 15 exons and generates a protein of 300 Kd size. APC protein has been identified, by immunoprecipitation methods, as b catanins. They interact with extracelluler matrix proteins through cadherins. They also interact with intracellular catanins. Mutations in them cause’s disruption of inter cellular organization, because of this and secretion of certain proteases from transformed cells release the contact inhibition and cells acquire mobility. Just one mutation is not enough to produce colon cancer, which is found in 50% of the people who are above 70years. This carcinoma starts with APC mutation first in epithelial cells, here dye plastic foci develop; this may lead to early adenoma. Ras mutation adds more punch to already unstable cells to develop into adenoma. Further mutation or deletion of DCC and finally p53 and loss of cadherins leads to metastasis. It is cumulative process over long period of time, it is important for people to have good and smooth bowel movement.
Nuclear Transcription Factors:
There are a number of transcription factors (TFs), which act as oncogenic proteins. Several proto-oncogenes such as Rel (a member of NFkB family of genes), Jun and Fos (hetero-dimeric enhancer binding proteins), erb-A (a nuclear localized receptor cum transcription factor of Thyroxin and other hormone family member), myc, and myb code for transcriptional factors and they are expressed at early stages of mitogen activation of cells in Go stage and they are involved in a cascade of events in cell cycle. On the contrary if they are activated inappropriately they can cause cell cycle go haywire. Similarly several onco-gene products also act as transcription factors, which also change the expression of several genes involved in cell cycle progression. Such action of TFs is manifested in several populations of mRNAs, which can be measured and compared with tumor and non-tumor cells, which provides intrinsic knowledge of molecular mechanisms and events that takes place with in cells, transformed or non-transformed.
Many nuclear Onco-proteins are expressed when normal and quiescent cells are induced by mitogens. For example 3T3 cells, which are quiescent cells, when treated with PDGF, induce the production of C-Fos and C-Myc by 50 fold. Initially C-Fos concentration dramatically increases and then it falls, but the production of C-Myc rises and remains prolonged. C-Fos and C-Myc are known stimulate transcription of genes encoding proteins that promote progression through G1 phase and G1 to S phase transition of the cell cycle. In tumor cells the expression of C-Fos and C-Myc and other genes is very high. Normal cells also express these mRNAs, but they are transient and degraded very fast.
Advances in the molecular biology of oncogenesis have established a key role for transcription factors in malignant transformation. In some cases the activity of the transcription factor itself is altered by mutation. In many other cases, the activity of the transcription factor is affected by mutations in upstream signaling or regulatory proteins. This review highlights four transcription factors--Stat3, Stat5, NF-kappa B, and HIF-1--which are associated with cancer development. The evidence for the involvement of these factors in oncogenesis is reviewed. Further, we examine the efforts to specifically target these transcription factors for therapeutic intervention. Such strategies include using peptidomimetics, antisense oligonucleotides, small molecule inhibitors, and G-quartet oligonucleotides. Inhibition of transcription factor activity may occur at the level of activation, translocation, or DNA binding. Application of these approaches to in vitro and in vivo models of tumorigenesis is going on.
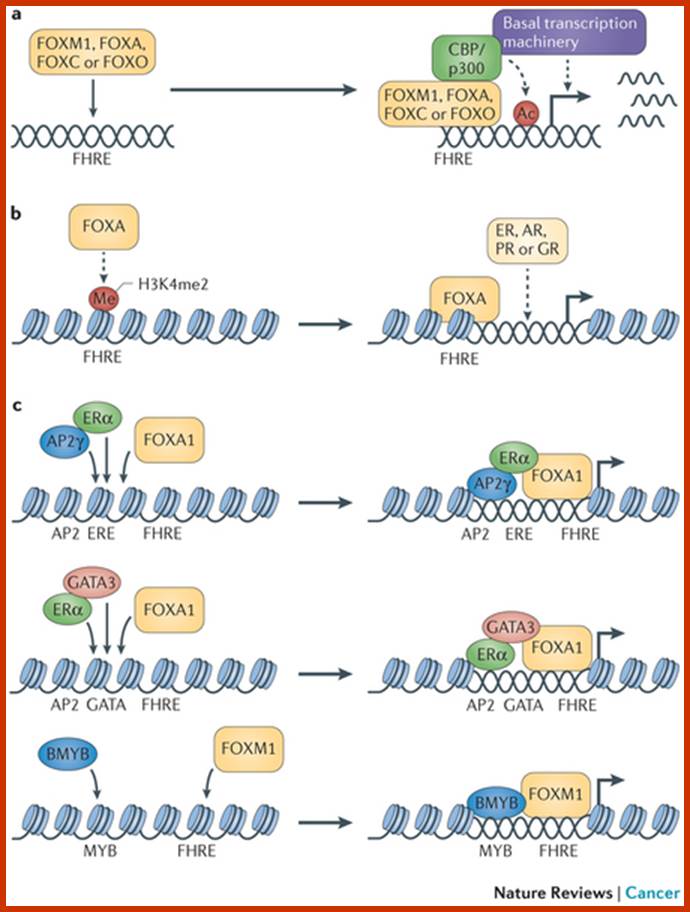
Forkhead box (FOX) proteins are multifaceted transcription factors that are responsible for fine-tuning the spatial and temporal expression of a broad range of genes both during development and in adult tissues. This function is engrained in their ability to integrate a multitude of cellular and environmental signals and to act with remarkable fidelity.
 and
and
http://www.nature.com/
Several key members of the FOXA, FOXC, FOXM, FOXO and FOXP subfamilies are strongly implicated in cancer, driving initiation, maintenance, progression and drug resistance. The functional complexities of FOX proteins are coming to light and have established these transcription factors as possible therapeutic targets and putative biomarkers for specific cancers. Eric W.-F. Lam etal; www.nature.com
V.Rel and NF-kB:
V-Rel causes Avian Reticulo endotheliosis. The virus has acquired Rel gene from normal cells. After viral infection the V-Rel is expressed causing lymphomas. This is due to the fact that the viral Rel has suffered a deletion of 100 amino acids at the C-end and also contains few point mutations elsewhere. V-rel is exclusively nuclear for its loss of c-terminal segment of the protein and it is highly oncogenic. Oncogenic Rel factors cause B-cell lymphomas. C-Rel has some 60% similarity in sequences with that of 50KD subunit of NF-kB. When V-rel, a truncated protein, as dimers with one of the cellular proteins remains in the nucleus and influence the gene expression either negatively or positively.
http://www.streetinsider.com/
Normal C-Rel belong to a family of genes coding for transcription factors, among them the best characterized one is NF-kB, a transcription factor. The NF-kBs are dimers of 65KD and 50KD and they are held in cytoplasm by I-kB. Binding of I-kB masks their DNA binding domain. When the I-kB is phosphorylated due mitogen activated kinases, the dimers are released from inhibition and now they enter the nucleus, where they bind to their respective promoters cum enhancer regions and activate specific gene transcription and soon the I-kB is degraded. NF-kBs are considered as one of the most pleotropic factors; they act as dimers and act as second messengers at nuclear level. Variety of stimuli result in activation of NF-kB and a broad range of genes are expressed; in appropriate expression of genes activated NFkB can lead to cell proliferation.
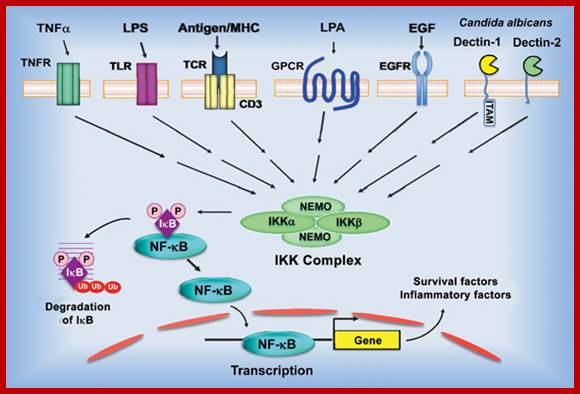
Blonska M, Lin X.NF-kappaB signaling pathways regulated by CARMA family of scaffold proteins. Cell Res 21:55-70
Model of NF-κB activation by the canonical pathway. Stimulation of the surface receptors by different inducers initiates several proximal signaling events resulting in activation of the IκB kinase (IKK) complex, composed of two kinases, IKKα and IKKβ, and the regulatory subunit NF-κB-essential modulator (NEMO). IKK phosphorylates inhibitor of κB (IκB), which leads to its ubiquitination and subsequent degradation. NF-κB is then translocated into nuclei and initiates the target gene transcription. TNFR – tumor necrosis factor receptor; TLR – toll like receptor, LPS – lipopolysaccharide; TCR – T-cell receptor; GPCR – G protein-coupled receptors; LPA - lysophosphatidic acid; EGFR - epidermal growth factor receptor; ITAM - immunoreceptor tyrosine-based activation motif; Ub - ubiquitin.
The NF-κB subunits RelA/p65, RelB, c-Rel, p50/p105 (NF-κB1) and p52/p100 (NF-κB2) comprise a family of dimeric transcription factors with both common and distinct biological functions. When the genes encoding NF-κB subunits were first isolated, their homology to the previously identified c-Rel proto-oncogene and its viral homologue v-Rel was clear. This provided the first indication that these transcription factors also had a role in cancer.
Rel/NF-![]() B transcription
factors are key regulators of immune, inflammatory and acute phase responses
and are also implicated in the control of cell proliferation and apoptosis.
Remarkable progress has been made in understanding the signal transduction
pathways that lead to the activation of Rel/NF-
B transcription
factors are key regulators of immune, inflammatory and acute phase responses
and are also implicated in the control of cell proliferation and apoptosis.
Remarkable progress has been made in understanding the signal transduction
pathways that lead to the activation of Rel/NF-![]() B factors and the
consequent induction of gene expression. Evidence linking deregulated Rel/NF-
B factors and the
consequent induction of gene expression. Evidence linking deregulated Rel/NF-![]() B activity to oncogenesis in mammalian systems has emerged in recent
years, consistent with the acute oncogenicity of the viral oncoprotein v-Rel in
animal models. Chromosomal amplification, overexpression and rearrangement of
genes coding for Rel/NF-
B activity to oncogenesis in mammalian systems has emerged in recent
years, consistent with the acute oncogenicity of the viral oncoprotein v-Rel in
animal models. Chromosomal amplification, overexpression and rearrangement of
genes coding for Rel/NF-![]() B factors have
been noted in many human hematopoietic and solid tumors. Persistent nuclear NF-
B factors have
been noted in many human hematopoietic and solid tumors. Persistent nuclear NF-![]() B activity was also described in several human cancer cell types, as
a result of constitutive activation of upstream signaling kinases or mutations
inactivating inhibitory I
B activity was also described in several human cancer cell types, as
a result of constitutive activation of upstream signaling kinases or mutations
inactivating inhibitory I![]() B subunits.
Studies point to a correlation between the activation of cellular gene
expression by Rel/NF-
B subunits.
Studies point to a correlation between the activation of cellular gene
expression by Rel/NF-![]() B factors and
their participation in the malignant process. Experiments implicating NF-
B factors and
their participation in the malignant process. Experiments implicating NF-![]() B in the control of the apoptotic response also support a role in
oncogenesis and in the resistance of tumor cells to chemotherapy. This review
focuses on the status of the rel, nfkb andikb genes and their activity in human
tumors and their association with the onset or progression of malignancies. http://www.nature.com/
B in the control of the apoptotic response also support a role in
oncogenesis and in the resistance of tumor cells to chemotherapy. This review
focuses on the status of the rel, nfkb andikb genes and their activity in human
tumors and their association with the onset or progression of malignancies. http://www.nature.com/
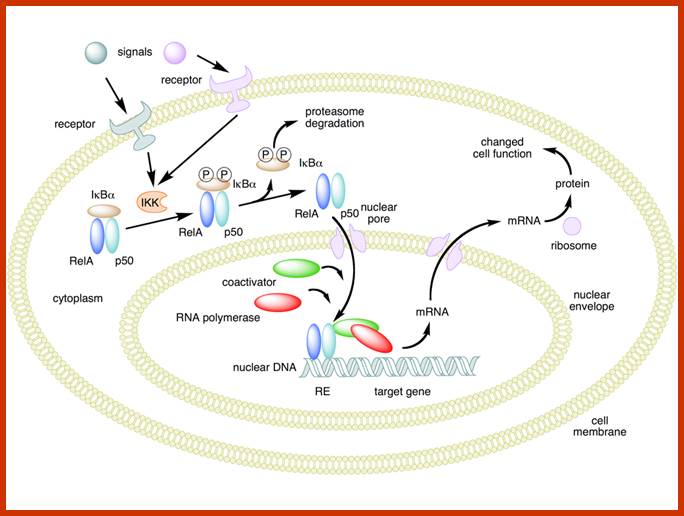
www.wikipedia.org; Nature Review-Molecular Cell Biology


NFKB1(top), RELA (bottom)
Mechanism of NF-κB action. In this figure, the NF-κB heterodimer between Rel and p50 proteins is used as an example. While in an inactivated state, NF-κB is located in the cytosol complexed with the inhibitory protein IκBα. Through the intermediacy of integral membrane receptors, a variety of extracellular signals can activate the enzyme IκB kinase (IKK). IKK, in turn, phosphorylates the IκBα protein, which results in ubiquitination, dissociation of IκBα from NF-κB, and eventual degradation of IκBα by the proteosome. The activated NF-κB is then translocated into the nucleus where it binds to specific sequences of DNA called response elements (RE). The DNA/NF-κB complex then recruits other proteins such as coactivators and RNA polymerase, which transcribe downstream DNA into mRNA, which, in turn, is translated into protein, which results in a change of cell function; https://en.wikipedia.org
AP1 Family of TFs: AP1 are a group of nuclear transcription factor that bind to enhancer region of upstream regulator sequences.
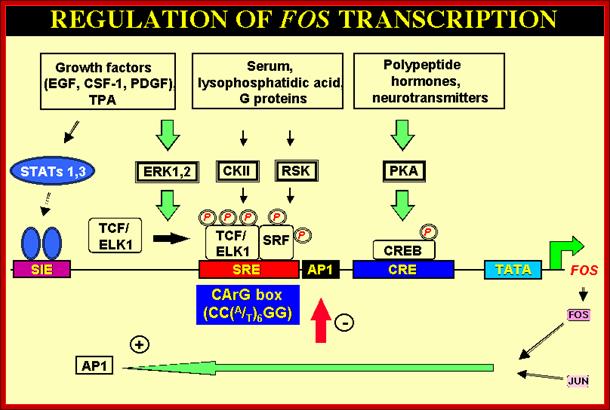
mol.cell.biol
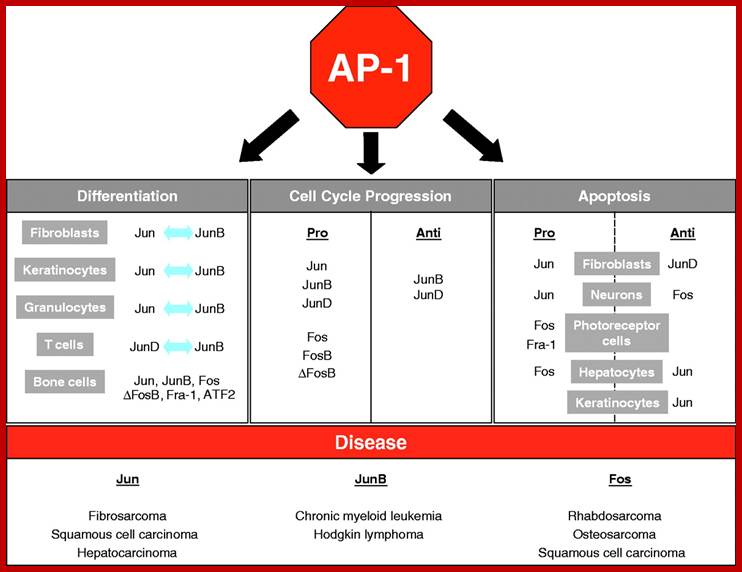
The AP-1 transcription factor is mainly composed of Jun, Fos and ATF protein dimers. It mediates gene regulation in response to a plethora of physiological and pathological stimuli, including cytokines, growth factors, stress signals, bacterial and viral infections, as well as oncogenic stimuli. Studies in genetically modified mice and cells have highlighted a crucial role for AP-1 in a variety of cellular events involved in normal development or neoplastic transformation causing cancer. However, emerging evidence indicates that the contribution of AP-1 to determination of cell fates critically depends on the relative abundance of AP-1 subunits, the composition of AP-1 dimers, the quality of stimulus, the cell type and the cellular environment. Therefore, AP-1-mediated regulation of processes such as proliferation, differentiation, apoptosis and transformation should be considered within the context of a complex dynamic network of signalling pathways and other nuclear factors that respond simultaneously; http://jcs.biologists.org/
Some are involved in a Phorbal ester (TPA a carcinogen) induced tumor formation. AP1 consists of dimer subunits coded by c-jun and c-fos. C-jun and c-fos are Leucine zipper proteins. They act in pair wise manner and generate a variety of TFs specific-to-specific class of genes. They activate genes whose promoters and enhancers have AP1 binding sites (enhancer sequences). Transcription factors encoded by C-Jun and C-Fos associate to form hetero dimeric TFs called AP1. They can also act independent of each other. Such genes were found in many transforming Retro viruses. The same genes were also found over expressed in many human tumors. So they can act as onco-proteins for they bind to promoters or enhancer regions of certain genes, whose expression cause expression growth promoting proteins or growth inhibiting proteins. The C-jun is activated by phosphorylation at two serine sites by JNK kinase. JNK kinase is actually activated by Ras pathway. The V-jun is a truncated version of C-jun (deletion of few amino acids) and it lacks site for JNK mediated phosphorylation, so it is not affected by ras pathway. Some other changes in C-jun make it constitutively active. Mutation in these genes abolishes transformation activity. This is a par excellent example for specific gene expression for cell transformation.
Colon Cancer:
Mutation in APC (chromosome 5)>
Activation of K Ras Oncogene (chromosome 12) >
Loss of DCC tumor suppressor gene (chromosome 18)>
Loss of P53 tumor suppressor gene (chromosome 17)>
Mutation in few more genes>
Carcinoma>
Human beings with one of the alleles disabled will be predisposed for cancer.
C-Erb A: This gene codes for a thyroid hormone receptor, which is member of steroid hormone receptor family. Binding of the ligand to the receptor leads to the binding of the complex to specific promoter elements or response elements, probably with co activators activate specific genes. Receptors are located in the nucleus and they are bound to specific response elements of DNA but inactive; however when the ligand tri iodo thyroxine binds, the receptor becomes active and stimulates gene expression. C-erbA- binds to tri-iodo-thyroxine with high affinity, and activates the expression of genes for suppression cell transformation, but allow cell differentiation. On the contrary the mutated gene V-erbA gene product does not bind to the hormone in mammalian cells. So V-erbA blocks the (rendered as a dominant negative Oncogene) suppressor and differentiation activities of C-Erb-A and allows cell proliferation instead of differentiation. So v-erbA contributes to cancer by blocking differentiation of erythroblasts that are transformed by v-erb
Myc Gene:
Over expression of causes cancers like Bursa lymphoma, T-cell lymphoma due to Avian leukemia viral and Murine leukemia viral infection respectively. Myc gene product is a transcriptional factor, which binds to promoters of those genes involved in controlling checkpoints in cell cycle. But over expression of such genes can be deleterious. Interestingly over expression of just Myc gene in transgenic mice doesn’t cause cancer immediately but it takes a long time say 100 days in only few mice. Expression of Ras D alone causes cancer in 150 days in 50% of mice. But if both Ras and Myc are expressed cancer develops very early and in most of the mice, however they require some more bad genes.
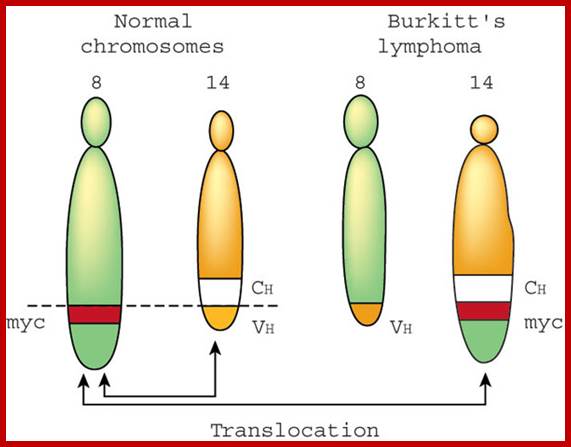
Reciprocal chromosomal translocations in Burkitt's lymphoma, a solid tumour of B lymphocytes; The genes for making the heavy chains of antibodies (Ch) are located on chromosomes 14, whereas those for making the light chains are on chromosomes 2 and 22. These genes are expressed exclusively in B lymphocytes, because only these cells have the necessary transcription factors to switch on their expression. In most (over 90%) of Burkett’s lymphoma cases, a reciprocal translocation moves the proto-oncogene c-myc from its normal position on chromosome 8 to a location close to the antibody heavy-chain genes on chromosome 14 (ref. 18). In other cases, c-myc is translocated close to the antibody genes on chromosome 2 or 22. In every case, c-myc now finds itself in a region of active gene transcription, and it may simply be the overproduction of the c-myc product (a transcription factor essential for cell division) that propels the lymphocyte down the pathway towards cancer. http://www.nature.com/
Myc gene perse due to certain point mutations, it can turn into cancer causing genes. The longevity of Myc protein is transitory and they are degraded by ubiquitination method. Ubiquitination is site specific. Often one finds a series of mutation in the N-terminal region ubiquitination sites are lost, thus the protein is not degraded and persist; this can activate several genes for the progression of Cell cycle events ceaselessly.
Under in vitro conditions, with low concentration of Epidermal Growth Factor or PDGF, cells get blocked at GO or G1 stage. Recombinant cells over expressing Myc in culture conditions; instead of going into cell cycle mode they die by Apoptosis. This is because cells receive inappropriate signals from Myc in the absence of other growth signals from the surface receptor and commit apoptosis. But in such conditions over expression of Bcl-2 (Breast cancer 2 genes), inhibits apoptosis and rescues cells from Myc mediated cell death, resulting cell proliferation. This happens even in the absence of normal growth factors. In Leukemia and Lymphomas both Myc and Bcl-2 genes over expressed.
This Myc gene that produces transcriptional factor activates genes required for passage through G1 to S-phase checkpoint and also for S-phase DNA replication. Over expression of this gene can deregulate cell division. And cells proliferate without any regulation. Integration of Avian viral DNA bordering the promoter of Myc gene, activate the Myc gene by virtue of its viral enhancer region, which is next to the Myc, such that Myc gene is over transcribed. Actually expression of Myc is down regulated in differentiated cells, but not in this case; the LTR-enhancer driven expression does not respond to any signals.
Such expressions are also found in mouse and other systems. In these tumors are found in antibody producing cells. Antibody producing calls are prone to make mistakes for they perform somatic recombination during production of Immunoglobulin. Here the myc gene is inserted at the enhancer region of the gene encoding Immunoglobulin Heavy chain.
Other genes such as C-erb B, C-myc, C-mos, R-raf and nearly another 10 genes are also activated and expressed in similar fashion but the enhancer is retroviral Viral LTR-Enhancer.
RNA Viral Onco Genes:
Retroviruses (RNA viruses) carry host proto-oncogenes from the previous infection, by a process called transduction (similar to Phage transduction of bacterial genes). These genes may be oncogenic or normal. If the gene they carry happens to be an oncogene, on infection, they cause cancer by the mechanism, as any other oncogenes are capable. Hitherto, description of genes and their oncogenic mechanism given is of C-oncogenes.
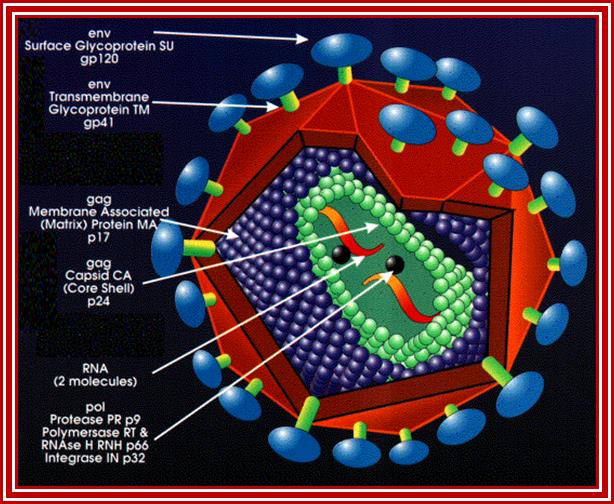
http://www.evolutionarymodel.com/

Retroviral oncogenes: 50 years of discovery; Peter K. Vogt. http://www.nature.com/
Retroviruses are the original source of oncogenes. The discovery and characterization of these genes was made possible by the introduction of quantitative cell biological and molecular techniques for the study of tumour viruses. Key features of all retroviral oncogenes were first identified in src, the oncogene of Rous sarcoma virus. These include non-involvement in viral replication, coding for a single protein and cellular origin. The MYC, RAS and ERBB oncogenes quickly followed SRC, and these together with PI3K are now recognized as crucial driving forces in human cancer. Peter K. Vogt

https://web.stanford.edu/

http://www.genetherapyreview.com/
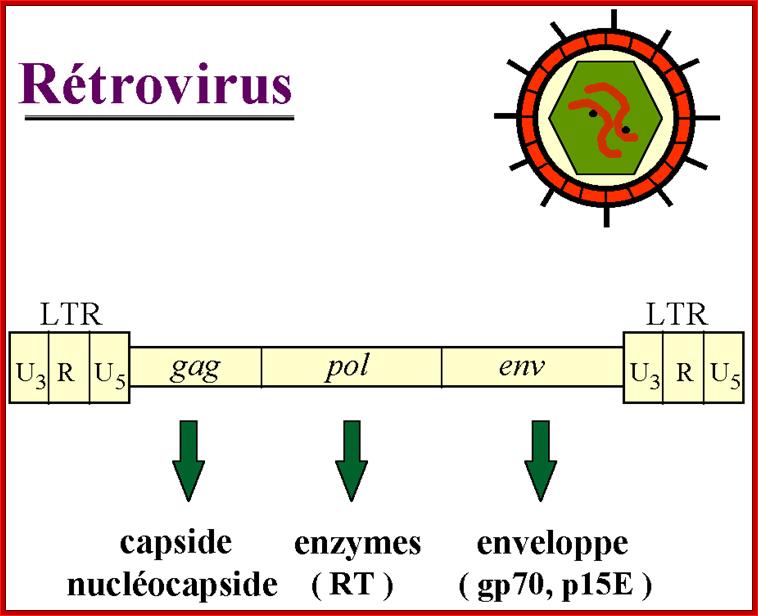
Retroviruses and their genomes:
|
Acronyms |
Names |
Year- by author |
Size and number |
Disease |
RSV |
Rous sarcoma virus |
Peyton- 1911 |
8kb RNA |
Cancer in chick and monkeys |
|
ALV |
Avian Myeloblastasis virus |
|
10kb |
Leukemia in birds |
|
ALV |
Avian Erythroblastasis virus |
|
10kb |
Leukemia in birds |
|
MMTV |
Mouse mammary tumor virus |
|
|
Mammary tumors in mice and monkeys |
|
MuMLV |
Maloney Murine leukemia virus |
|
|
Mice and cats |
|
HTLV 1 |
Human t-cell leukemia virus-1 |
Robert Gallow-1981 |
9.5 kb |
Transform CD4 –T-lymphocytes |
|
HTLV-2 BLV-HTLV |
Human T-cell transforming virus |
Robert Gallow |
9.5kb |
CD4-T cell transformation |
|
HSRV |
Human Spuma virus |
|
|
Human cell transformation |
|
MPMV |
Mason Pfizer Monkey virus |
|
|
|
|
HIV 1 |
Human immunodeficiency virus |
1983 |
9kb |
Immune deficiency |
|
HIV 2 |
HIV-2 |
|
9kb |
Helper T-cells deformed |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Few Cancer Genes carried by Retroviruses:
|
Virus |
Host |
Disease |
V-Gene |
|
RSV, Rous sarcoma |
Chick |
Sarcoma |
V-Src |
|
HaMUSV Harvey murine sarcoma |
Rat |
Sarcoma & erythro leukemia |
V-H-ras |
|
Ki-MuSV, Kirsten murine sarcoma |
Rat |
Sarcoma |
V-K-ras |
|
Mo-MuSV, Moloney Murine sarcoma |
Mouse |
Sarcoma |
V-mos |
|
FBJ-Musv, FBJ Murine osteosarcoma |
Mouse |
Chondrosoma |
V-fos |
|
SSV simian sarcoma |
Monkey |
Sarcoma |
V-Sis |
|
PiFe SV, Feline sarcoma |
Cat |
Sarcoma |
V-fes |
|
Sm-Fesv, Feline fibrosarcoma |
Cat |
Fibrosarcoma |
V-fms |
|
ASV-17, Avian sarcoma |
Chick |
Fibrosarcoma |
V-jun |
|
FuSV,fujinami sarcoma |
Chick |
Sarcoma |
V-fps |
|
Myc 29, Myelocytomatosis |
Chick |
Myelocytoma, carcinoma, sarcoma |
V-Myc |
|
Mulv, abelson leukemia |
Mouse |
B-cell lymphoma |
V-abl |
|
Rev-T, reticuloendotheliosis |
Turkey |
Lymphatic leukemia |
V-rel |
|
AEV, avian erythroblastosis |
Chick |
Erythroleukemia, fibrosarcoma |
V-Erb B-A |
|
AMV, Avian myeloblastosis |
Chick |
Meyloblastsic leukemia |
V-Myb |
|
|
|
|
|
Note: Here V-oncogenes mean, the viruses that carry host genes by transduction and they are not of viral genes, these V-genes can cause cancer. All the mentioned genes are host cell genes; they are called proto-oncogenes for they have the potency to change into oncogenes.
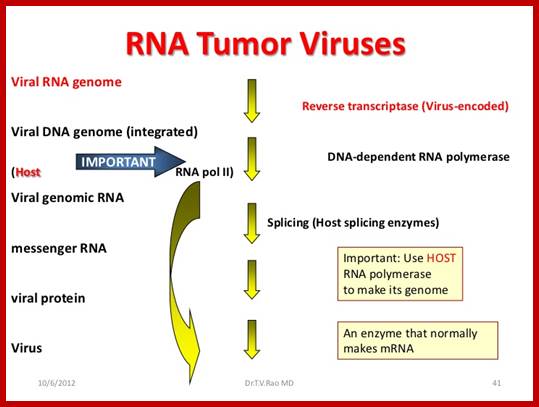
 -
-
http://www.lookfordiagnosis.com/
Retroviruses carry pair identical RNA genomes, which on infection the capsid is released into the cytoplasm. The viral RNA is positive RNA and contains all the coded information for translation, has 5’ cap and 3’ polyA-tail, but the RNA is not translated for it is not released from the capsid. As the capsid carries the required enzymes and factors, the RNA genome is transcribed as complementary DNA called cDNA. The replicated cDNA is double stranded but larger than the original size of the viral RNA. This because each of the ends contains U3, R5 and U5 in the same order at one end and at the other ends it has U3, R3 and U5 in the same order. These segments at either end are called Long Terminal Repeats (LTRs). The U3 at the proximal end has promoter and enhancer sequences. In general the viral carries Gag, Pol and Env genes in the same order, but many viral genomes contain some additional genes especially between Pol and Env genes e.g. HIV and HTLV genomes. Once the genome is replicated the ds cDNA circularizes because of the binding of integration proteins, this circular form is transported into the nucleus, where it is integrated into the host genome. The site of integration is random and sequence bias. In this form it can remain for any number of days or life long, if it happens to be integrated in germ line cells, the viral DNA can be vertically transmitted.
Random integration at sites has significant consequences. If the viral genome integrates in the upstream of cellular gene such as Myc or Ras, as the viral cDNA has promoter –enhancer regions, if the cellular genes present next to such elements, it is expressed continuously. This has an effect similar to other genes that cause cancer. Major number of viral induced cancer is mainly due to viral genome integration.
On the contrary the integrated viral genome when activated by certain factors, it transcribes, during this process, it can extend its transcription that includes a cellular gene if it happens to be at its end; this depends upon how and where the viral genome is integrated. When viral particle are formed, they will be carrying host cell gene in modified (mostly) form. Such genomes when on infection, if they are integrated, the cellular gene is expressed because of the promoter enhancer element of the viral genome. If the stolen gene happens to be a kinase, then the kinase is over expressed and that causes the tumorigenesis. For example Src gene carried by RSV is truncated by 19 amino acids at the carboxyl end, thus the expressed protein remains ever active and the gene is also expressed continuously, thus causes cancer disease.
DNA Viral Oncogenes:
Viruses, with DNA as the genome, on infection of permissible host cells, (monkey cell for SV 40), they multiply and cause lyses of cells, but on the contrary if they infect non-permissible cells (Rabbit cell for SV40), they don’t multiply, instead few among many thousands of viral particles, transfer their genetic material into host chromosomal DNA. Unlike retroviral v-oncogenes, the DNA viruses carry their own genes, which are capable inducing cancer. If the integrated viral genetic material has oncogenic property; it will transform cells into tumor cell lines. Majority of the DNA viral genes act against RB and p53 genes, thus they release cells from tumor suppressor activity.
|
Virus |
Genome |
Size (kip) |
Origin of onco-g |
Oncogene |
Action |
|
Polyoma/SV40 |
dsDNA |
5-6 |
Early viral gene |
T-antigen |
Inactivates tumor suppressor |
|
HPV |
dsDNA |
~8 |
Early gene |
E6 and E7 |
Inactivates tumor suppressor suppressor |
|
Adeno |
dsDNA |
37-38 |
Early |
E1A & E1B |
Inactivates tumor suppressor |
|
EPBV (Epstein) |
ds DNA |
|
Early |
|
B-Cell transformation, lymphoma |
|
BKV |
|
|
|
|
Burkit’s lymphoma |
|
|
|
|
|
|
|
Note: Here viral c-oncogenes means the oncogenes belong to viral genome; they are not transduced host cellular genes. They carry their own genes, when they integrate into host genome; they cause cancer by suppressing the activity of tumor suppressors.
RNA Viral C-Oncogenes:
|
HIV |
ssRNA |
7-9 kb |
|
|
Incapacitates T-cell immune system |
|
HTLV |
ssRNA |
7-8kb |
|
|
Incapacitates T-cell immune system |
|
MMTV |
ssRNA |
6-7kb |
|
|
Mouse mammary tumor |
Note: Here viral c-oncogenes means, the proto-oncogenes belong to viral genome, they are not host cellular genes, but by inserting their genes into host chromosomes they cause oncogenesis.
Papovo viruses: Polyoma & SV 40 viruses:
Polyoma viruses contain ds DNA of 5-6Kbp sizes. At early stages of infection it produces a protein called T antigen. This is required for DNA replication and also for activation of late gene products. The SV 40 virus also produces another smaller version of antigen protein called ‘t’ antigen. Similarly Polyoma viruses generate three antigens, ‘T’ small “t” and middle T. they are the products of alternate splicing. The T-antigen binds to p53 and RB proteins, which are tumor suppressors. Thus it facilitates tumorigenic activity. The T-antigen is responsible for transformation and the small ‘t’ is responsible for the maintenance of the transformation. But Polyoma T causes cell immortalization and middle T is responsible for transformation. The T and middle T cause tumors.
Human Papilloma virus:
Human Papilloma virus (HPV) contains a ds DNA, of 8 Kbp long. It transcribes early genes and the products are E6 and E7, inactivate TSG. Human Papilloma infects epithelial cells and cause epithelial cancers. E6/E7 proteins are the causative factors and bind to tumor suppressor proteins and inactivate them, thus they cause immortalization of cells. But the E5 dimerizes and binds to cytosolic PDGF receptor and cause sustained activation of receptors. The virus causes skin and genital warts

https://lookfordiagnosis.com
Adeno virus:
Adeno virus, (dsDNA-38Kbp) infects respiratory system; express certain genes very early on infection; they are called E1A and E1B. E1A and E1B are the causative factors. They activate few genes and repress few genes. E1A transcript consists of 4 exons (1,2,3 and 4) with introns. It is alternatively spliced to generate three proteins of 289 (all exons are included), 243 (exons 1,2 and 4 are included) and 55 (only 1 and 4th exons are included) amino acids long.
The early expressed gene products E1A have the ability to immortalize cells and can cooperate with other onco proteins such as Ras and transform cells. For transcriptional activation the protein generated should have exon domain 3, without that it cannot activate host gene activation. Domain 1 and 2 are required for repression of transcription, induction of DNA synthesis and morphological transformation.
E1A targets co activators like CBP and p300, TBP and cell cycle regulators like RB and p27. E1A binds to RBA and releases E2F transcriptional factors required for cell cycle progression events. E1A also binds to p27 and releases Cdk-Cyclins from blockage. E1A also represses certain cellular genes.
CAR receptor-fiber-knob Ad interaction and internalization process; https://microbewiki.kenyon.edu
RetinoblastomaE1B binds to p53 and inactivates its apoptotic domain; it also binds to transactivation domain and inactivates p53-mediated transcriptional activation of specific genes. E1B also activates several genes involved in cell proliferation
E1A with middle T of Polyoma virus can easily transform primary cell with efficiency. On the other hand E1B causes morphological transformations characteristic of cancer. E1A also represses certain cellular genes, thus tumorigenesis requires repression of certain cellular genes. It also activates cellular genes for cell transformation and cell proliferation. E4 acts as a death factor via Src.
Few more Viral Oncogenes:
FJB Murine Osseo sarcoma- causative gene-fos.
RSV- chick; the gene src.
Harvey murine SV- rat, Ha MuSV; gene-H-ras.
Kirsten murine sarcoma- Ki-Musv;gene- K-ras.
Maloney Murine Sarcoma- MoMuSV, gene- mos.
Simian sarcoma- SSV; gene-sis.
Feline sarcoma-PI-Fe-Sv; cat sarcoma, gene sis.
SM-Fe-S, Cat, fibrosarcoma; gene-fms.
St-Fe-Vs., cat, fibrosarcoma, gene fes.
Avian sarcoma, ASV-17, in chick fibrosarcoma, gene- jun.
Avian Myelocytomatosis, carcinoma, carcinoma and Myelocytoma; gene-Mc29 and myc.
Abelson leukemia, MuMLV, mouse B-cell lymphoma, gene- abl.
Avian Myeloblastasis, AMV, Myeloblastic leukemia, gene- myb.
Avian erythroblastasis, AEV, chick erythroleukemia, gene erb-b and erb-A
Retroviruses of Avian, Mouse, and Monkeys others, can activate cellular genes by inserting next to them. These viruses have LTR sequences, which contain promoter enhancer sequences, thus cellular genes get activated towards oncogenic pathway. The viruses and their genes can be transferred vertically and horizontally as well
Similarly a host of retroviruses carry oncogenic genes and on their infection they are found to cause tumors. All most all-oncogenic viruses carry host cellular genes, but they are modified in such a way, on entry into host cells, they get activated in one form or the other and cause cancer. The genes they have acquired are mutants where they have gained a function or lost a function, which causes proliferation of cells leading tumors. But HIV is an exception, which spreads and disables all T-cells. As a result variety of diseases develops including few cancers. HTLV is lymphotrophic.
Tumor Suppressor Genes:
Loss of PDGFβ Signaling:
Loss of PDGFβ (Tumor cell derived growth factor Beta) signaling contributes abnormal cell proliferation and malignancy. These factors are also secreted by most of the body cells as a defense against abnormal cell proliferation in a differentiated tissue and they have varied range of biological activities. But most important of them is their ability to inhibit the proliferation of many cell types including most of the epithelial and immune cells. Loss of PDGFβ contributes to the development of a variety of tumors.
The BMP proteins are homolog of PDGFβs, and they control many important developmental steps. PDGFβs signals sequential activation of two cell surface receptors; type-I and Type-II, both have intrinsic serine and threonine protein kinase activity. Binding of PDGFβ ligands to the receptors induces phosphorylation and activation of Type-I receptors by Type-II receptor kinases.
In this signal transduction process S-mad protein play an important role as intracellular signal transducers in the pathway down stream from the receptors to nuclear transcription. Activated Type-I receptors phosphorylate serine residues at the C-terminal region of either S-mad2 or S-mad 3, which enables them to bind to one or more molecules of S-mad4. S-mad4 is a common partner of all phosphorylated S-mads involved in signaling process by both TGFβ and bone morphogenic proteins. S-mads acts as intracellular signal transducers enter cytoplasm into the nucleus.
Activated S-mad proteins activate transcription of a variety of genes. Among them one important gene that is activated by PDGFβ is P15 gene, the G1 cyclin kinase inhibitor that displaces P27 protein from CDK4 cyclin-D complex. Freed P27 binds and inhibit Cdk2-cyclin-E complex, which is required for the entry of cell into S-phase. Thus PDGFB inducing the expression of P15 causes cells to arrest at G1 stage.
Many tumors contain inactivating mutations in PGFB receptors or S-Mad genes are resistant to growth inhibition by PDGFβ. Human pancreatic cancers contain a deletion in S-Mad4. This gene originally was called DPC (deleted in pancreatic cancer). Retinoblastoma, colon cancer, hepatoma and some T and B cell malignancies are found to be unresponsive to PGFβ treatment. Loss of responsiveness is due to loss of Type-I and Type-II PDGFβ receptors. Mutations in S-Mad2 genes are also found in many types of cancers. PDGF β action can be blocked by two nuclear proteins such as Ski and Sno, but Smad protein interact with these nuclear proteins and deliver to ubiquitinated proteosome digestion, thus favor PGF β action of tumor growth suppression.
The PDGFβ signaling pathway also induces expression of many genes responsible for the synthesis of extra cellular matrix substances such as collagen and induces the expression of an inhibitor of plasminogen-activator. Synthesis of ECM proteins reinforces the cell-cell adhesion and restricts the movement of cancer cells from spreading. This PAI-1 is an inhibitor of serum protease that degrades many extra cellular matrix proteins. In the absence of PAI-inhibitor, proteases released into extra cellular space may contribute to metastasis, allowing tumor cells to escape through extra cellular matrix free region. So the plasminogen activator protease is inhibited by PA-I, thus ECM is not degraded. The protein P15 also activates a gene that encodes a cell cycle inhibitor.
PDGFβ bind to PDGFβ receptor>
Phosphorylation of Type-I Receptor.>
Type-I (p) phosphorylates S-Mad3.
S-mad-2/3-P-S-mad4 complex enter the Nucleus.>
Bind to PAI-1 promoter with TF-E3>
Activate the gene expression of PAI-1
[Inhibition]
Retinoblastoma Tumor Suppressor Genes:
Retinoblastoma is a hereditary disease, which is expressed at early child hood in if the child is homozygous recessive for this gene, even if the child is heterozygous there is a possibility of manifestation of the gene at little later stages. Molecular weight of RB is 105.5 KD.

Rb halts the cell cycle and releases its hold in response to cell gowth; http://cnx.org/contents
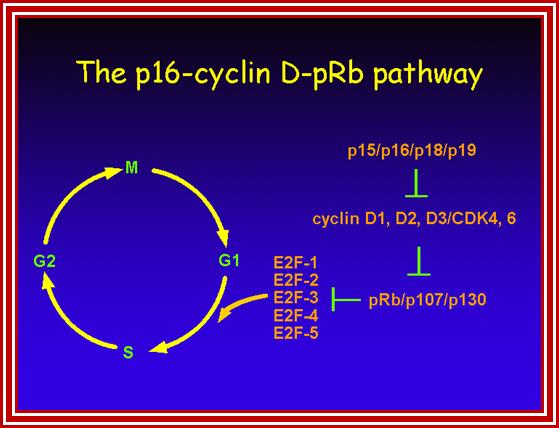
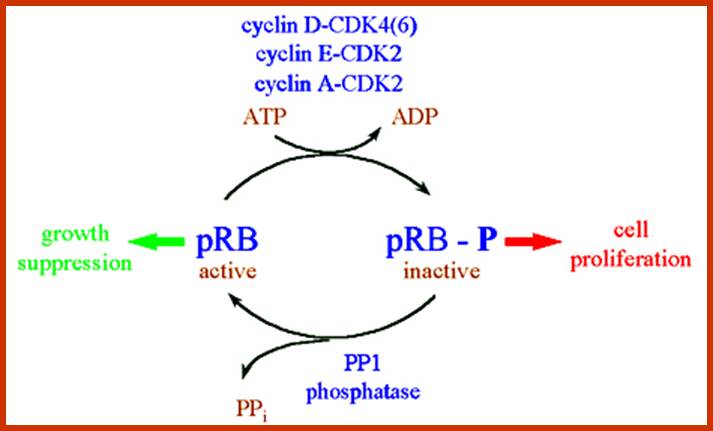 Frontiers in Bioscience3.;
Frontiers in Bioscience3.;
https://www.bioscience.org
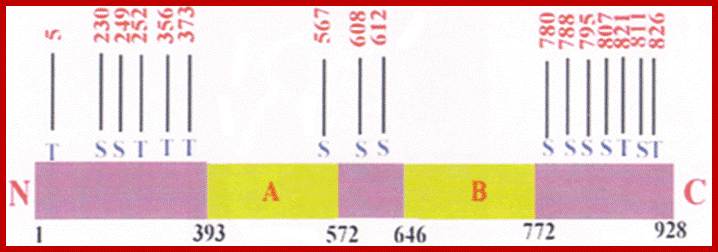
RB protein; http://annalsofneurosciences.org/
Rb gene consists of 27 exons- 200kb long, encodes 4.7kb mrna and 110 Rb protein; protein half-life is >8hrs. Protein has three structural domains, A and B are consistent among all species; it regulates cell entry into cell cycle, injection of RB1 protein prevents cell entry into cell cycle.

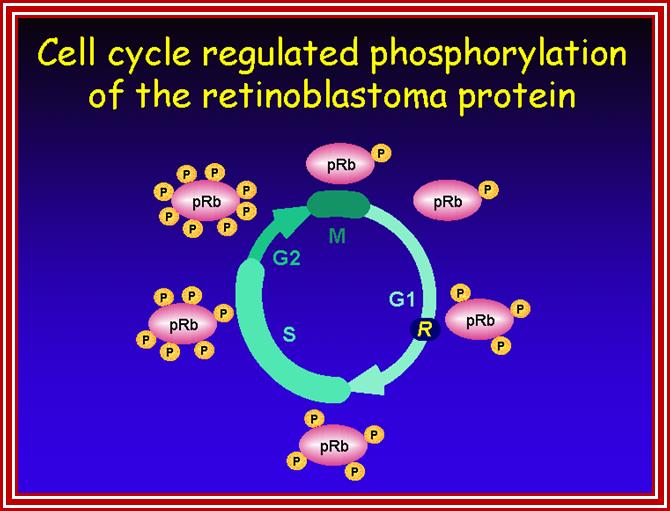
http://streaming.cineca.it/

pRB regulates cell cycle progression, differentiation and apoptosis. Hypophosphorylated pRB represses genes necessary for S-phase entrance by repressing transcriptional activation by E2F, causing cell cycle arrest, and activates genes necessary for differentiation by binding cell type-specific transcription factors. Phosphorylation of pRB stimulates exit from G0, and in late G1 phosphorylation by cyclin D-Cdk4/6 and cyclin E-Cdk2 kinases releases E2F, allowing cell cycle progression. Continual phosphorylation of pRB occurs by cyclin A-Cdk2 and cyclin B-Cdk1 complexes in S and G2 phases. pRB is activated in late mitosis when PP1 phosphatase dephosphorylates it. http://www3.unisi.it/ricerca
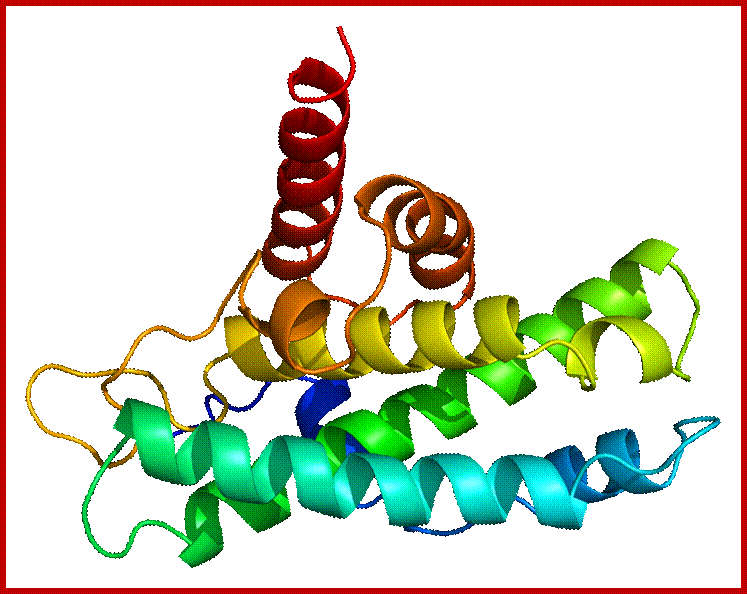
Retinoblastoma protein- www.wikipedia.org
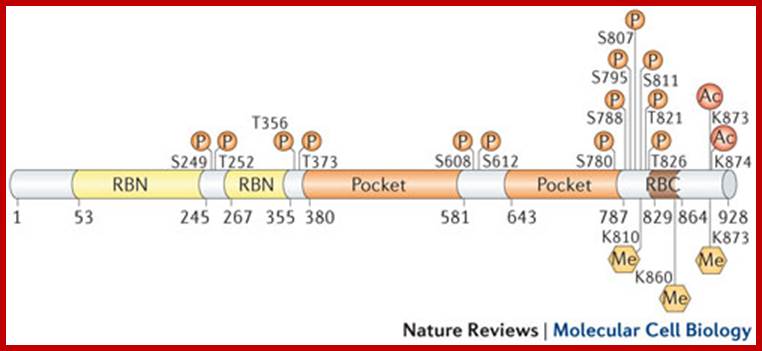
RB post translational modifications; Frederick A. Dick & Seth M. Rubin
http://www.nature.com/
Post-translational modifications have an important role in the regulation of RB function. With a few exceptions, RB phosphorylation (P) results in inactivation, transcriptional derepression and cell cycle progression. RB is phosphorylated by several different kinases, including cyclin-dependent kinases (CDKs) and checkpoint kinase 2 (CHK2). Phosphorylation controls RB interactions with other proteins. This modification typically occurs outside structured domains (see the figure) and promotes conformational transitions from disordered to ordered RB structures that mask protein-binding surfaces . Different kinases show preferences for particular phosphorylation sites, and discrete phosphorylation events induce specific structural changes. However, it remains uncertain whether, and in what context, differentially phosphorylated isoforms of RB exist in the cell.
Acetylation (Ac) and methylation (Me) sites have been identified in disordered sequences towards the RB carboxy-terminal domain (RBC)37. In contrast to phosphorylation, these modifications occur in response to signals, such as DNA damage and differentiation, which correlate with RB activation and repression of gene expression. Acetylation occurs on Lys873 and Lys874, which are located within the cyclin-docking sequence, and results in reduced phosphorylation, probably through kinase inhibition83, 84. Methylation on Lys873 and Lys810 by SET-domain methyltransferases similarly results in RB hypophosphorylation. SET and MYND domain-containing 2 (SMYD2) methylates Lys860, which results in the recruitment of the transcriptional repressor lethal(3)malignant brain tumour-like 1 (L3MBTL1). The reader is referred to other reviews for more details on these and other emerging post-translational modifications on RB and their roles in the regulation of function.
When the cell is in resting stage RB exists in unphosphorylated state. In this condition it binds to several E2F factors and prevent E2Fs activate gene products required for cell cycle events. E2F factors with RB in normal situation are bound to their respective gene promoter elements, and recruit histone deacetylases and cause inactivation of transcription. E2F factors are a family transcription factors. Thus RB acts as suppressor of cell division under inappropriate conditions. Mitogen activated cavalcade of kinase and kinase-kinase events, including Cdk-Cyclin D kinase activity, leads to phosphorylation of several components, one of them is RB also. Even Ras mediated kinase pathway also lead to phosphorylation of RB. When RB gets phosphorylated conformational changes in the protein releases all E2F factors.
The E2Fs (E2F1 to E2F6) are a family of transcription factors; function as heterodimeric proteins (DP). E2F factors are transcription activators or they may act as co activators too. They have DNA binding domain, but it is masked by the binding of RB Most of them when they are released act on specific promoters and initiate transcription genes, mostly of cell cycle progression factors from Go to S and G1 to S and S phase events. The genes expressed are related DNA synthesis, like DNA Pol, helicases and others, all required for progression of cell cycle through G1 to S and S-phase also.
It is not just phosphorylation events that release E2Fs from the surface of RBs; even the binding of SV40’s T-antigen and ADVs E1A protein inactivates RB from holding E2F factors., HPV’s E7 binds to RB, polyoma’s T-antigen binds to p53, all these make free RB from holding on to transcription factors. Deletion RB induces cancer such as osteosarcoma. If such osteosarcoma cells in cell culture are transfected with RB protein, cells atop growing. Even over expression cyclin-D induces cancer for it facilitates the cells enter into cell cycle mode. However there are few more protein factors like p107 and p130 that sequester E2Fs. They also release E2Fs on getting phosphorylated. Several regulators such as p16, p21 and p27 ensure inactivation Cdk-cyclin complexes. P16 blocks cdk4-cyclin D2, Cdk4-cyclinD1 and Cdk6-cyclin E. similarly p15, p16, and p18 and p19, bind to Cdk4-cyclin D2, thus prevent cell cycle progression into cell cycle. But losses of these proteins lead to uncontrolled cell division.
Wild type P53-Tumor Suppressor gene:
Among all tumor suppressor genes, wild type WTp53 is the most powerful regulator that acts at various stages of cell cycle to suppress tumor induction. P53 is named after its molecular weight 53Kd; it is Phospho-protein always found in the nucleus, becomes tetramer and acts. If one of the tetramer is damaged, the tetramer fails to function, which amounts to loss of function with characteristic dominant negative mutation. It’s half-life is very short-20 minutes. Its concentration in normal cells is low, but when cell suffers damage at DNA level the protein gets activated and become stable and also it concentration increases. If there is damage to DNA at G1 stage, p53 blocks cell progression beyond G1 stage. If DNA is damaged or replication produces any errors, again the progression of cell cycle is blocked at G2 stage. If the damage is beyond repair, p53 induces Apoptosis.

Interaction of p53 with immune system ;http://www.biodiscoveryjournal.co.uk/
P53 has many domains each of which has specific function; very rarely one finds a protein contains so many domains and so many functions. That is one of the reasons why animal systems including human beings, with mutations in p53, are generally susceptible to cancer.

P53 and NF-kB signaling in senescence and SASP; http://www.biodiscoveryjournal.co.uk/
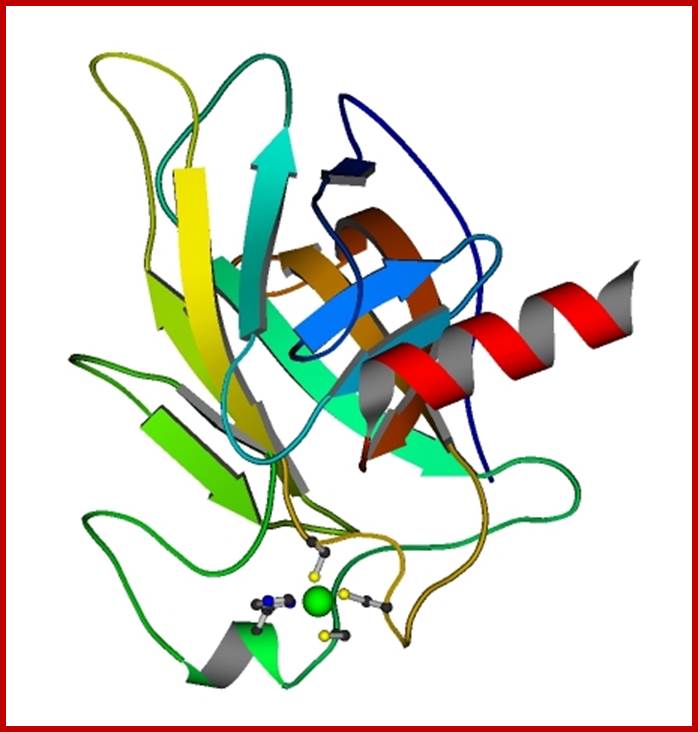
The p53 tumor suppressor protein; http://www.mrc-lmb.cam.ac.uk/
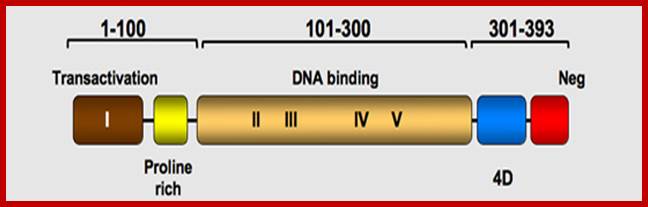

Human p53 protein (Hp53) can be divided into five domains, each corresponding to specific functions:; http://p53.free.fr/p53_info/p53_Protein.html
I) The amino-terminus part 1-42 contains the acidic transactivation
domain and the mdm2 protein binding site. It also contains the Highly Conserved
Domain I (HCD I)
II) Region 40-92 contains series repeated
proline residues that are conserved in the majority of p53. it also contains a
second transactivation domain.
III) The central region (101-306) contains the
DNA binding domain. It is the target of 90% of p53 mutations found in human
cancers. It contains HCD II to V.
IV) The oligomerization domain (307-355, 4D)
consists of a beta-strand, followed by an alpha-helix necessary for
dimerization, as p53 is composed of a dimer of two dimers. A nuclear export
signal (NES) is localized in this oligomerization domain.
V) The carboxy-terminus of p53 (356-393)
contains 3 nuclear localization signals (NLS) and a non-specific DNA binding
domain that binds to damaged DNA. This region is also involved in
downregulation of DNA binding of the central domain.
The amino-terminus of p53
AD1:
activation domain 1
AD2: activation domain 2
PRD: proline rich domain
NES: nuclear exclusion domain
HCD I: Highly Conserved Domain I
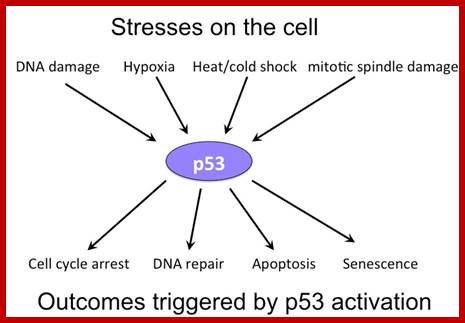
Cellular Morse Code; Actvation of p53 and its action; http://ethesis.helsinki.fi/; https://ittakes30.wordpress.com
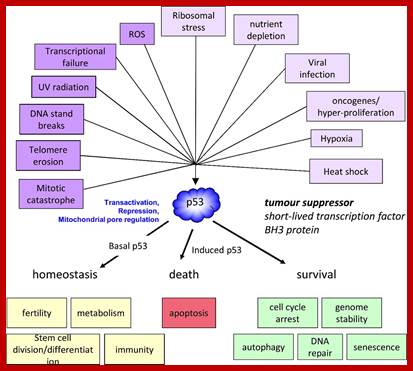
Regulation of the p53 response and its relationship to cancer; David W.Meek;http://www.biochemj.org/
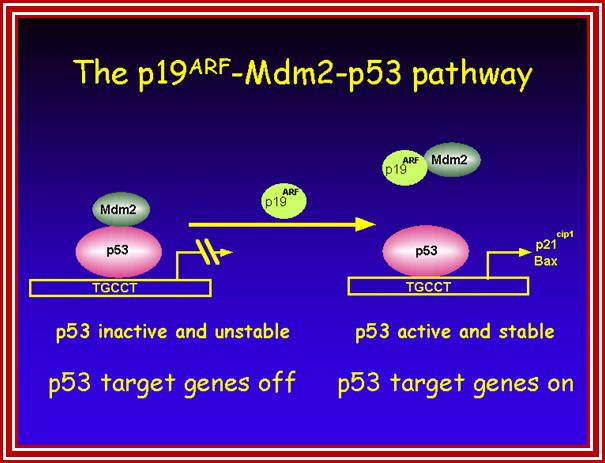
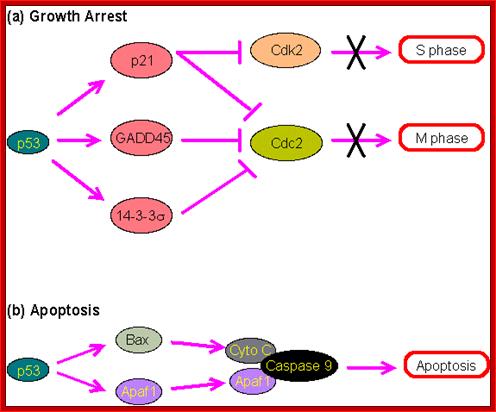
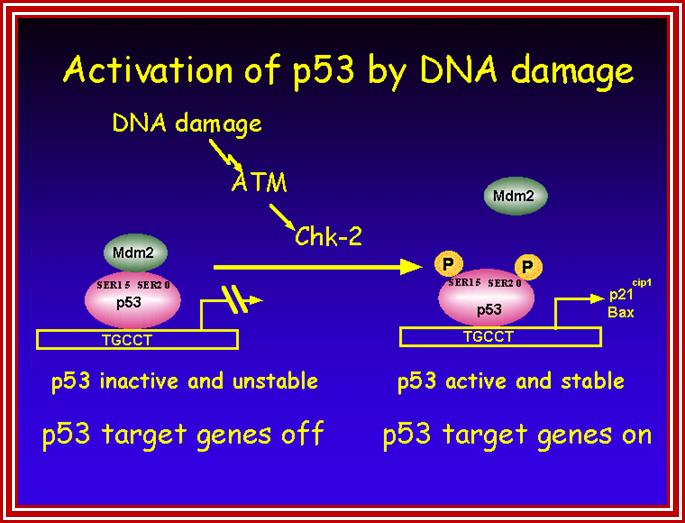
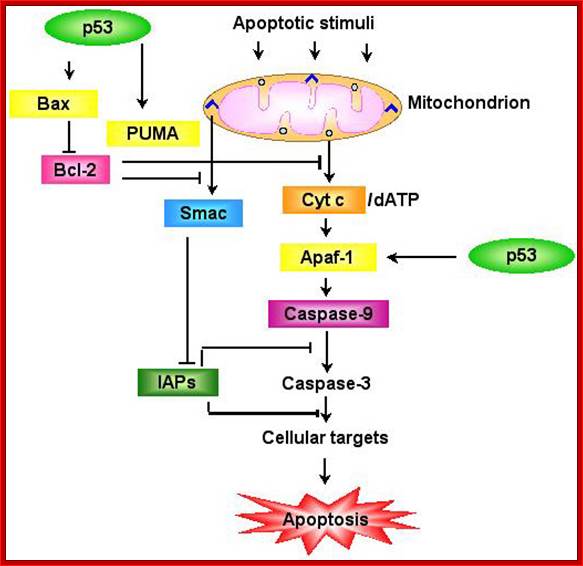
https://webhome.weizmann.ac.il
This protein acts like a vanguard among others against tumorigenesis. More than 50% of the cancer patients have p53 disfunctioned or disfunctioning p53 in mammary carcinoma. Mice homozygous recessive for p53 survive only for few months and 100% die, but mice with heterozygous live little longer and they are as good as dead, but statistics show 80% of them die. The protein perse has many domains.
· From N-end 1-50 aa it has transcription activation domain, interacts with TBP, mdm2 and coactivators-p300 & CBP. This region, from 43 to 63, contains domain for inducing apoptosis
· The protein domain between 50-100 contains a signaling domain, it is in the signaling domain, and the protein has DNA kinase, ATM kinase and ATR kinase motifs. In the signaling domain the sequence PXXP act as SH3 for protein-protein interaction.
· From 100 to 290 AA, it has palindromic 10 base pair long DNA binding domain; the p53 protein binds to such sequences in and activates respective genes. This is same region to which SV40 T-antigen also binds.
· At C-end between 320 and 350 the protein has oligomerization (tetramerization) domain.
· The segment between 350-390 AA binds damaged single stranded DNA with 3’ ends including Telomeric DNA. Binding of protein to 3’ ends activates the p53.
P53 is a nuclear Phospho protein. To perform any activities the p53 should be in tetrameric state where all the subunits should be structurally normal. Environ signals such as UV radiation caused breaks in DNA, with damaged single strands, Telomeric DNA activate its ATR kinase II (it is ATM related) domain, and ionizing radiation activates its ATM kinase (Ataxia Telangiectasiactasia) and make the p53 active and stable. The activated kinases phosphorylate the protein at serine 15, ser33 and ser392. Possibly its lysine residue is also acetylated (very rarely). To activate the N-terminal region of p53, which initiates transcription, the binding of p53 to DNA through the concensus palindromic sequences is very much essential.
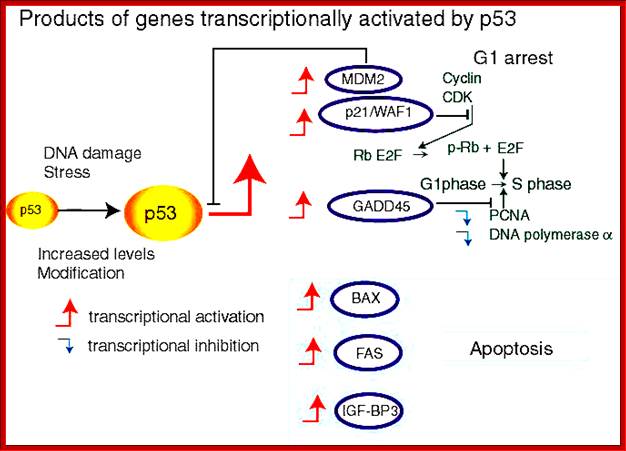
One of the earliest genes transcribed by p53, among others, is p21. The p21 protein is an inhibitor of Cdk2-Cyclin E, which prevents entry of cell cycle from G1 to S phase. Similarly activated p27 and p16 also block the action of Cdk4, 6-Cyclin-D’s cell cycle progression. The activated p53 also activates genes GADD45, for DNA repair that is damaged due to radiations. The N-terminal region acts as trans activator and interacts with TBP. It also uses p300 and CBP as trans activators.
P53 also trans activates few genes involved in apoptosis pathway. One of them activates surface receptors that triggers apoptotic pathway, like Fast receptor. Another interacts with mitochondrial membrane and release Cytochrome-C, which activates the whole series of Caspases and cause cell death.
With the damage to Telomeric DNA to a crisis level, cells enter into senescence stage. P53 responds to senescence and activates genes for growth arrest and also induces apoptosis. On the contrary Bcl2 when activated when activated because of translocation inhibits apoptosis and favors tumorigenesis. But p53 favors cell growth arrest and favors apoptosis, when cells genetic stability at greater risk. Thus p53-induced apoptosis is the major pathway in blocking tumorigenesis. At the same time p53 is essential for development.
Even, certain signals, emanated from abnormally growing cells, activate p19 ARF, which in turn inactivates mdm2 protein; thereby p53 remains active. Mdm2 is a regulatory protein; on activation it targets p53 for ubiquitinated proteosome mediated destruction of N-end of the protein, thus inhibits transactivation with p300 and Cpb. So mdm2 is an anti-suppressor protein. Deletion of p19ARF gene facilitates the movement off mdm2 into the nucleus and degrades p53, thus favors tumor induction. It is important to note that those proteins, which activate p53, are tumor suppressors and those that inactivate are tumor facilitators.
When p53 suffers mutation in one or the other form, cell at any time can to transformed into tumor with other mutated cancer causing genes.
· Mutation in the DNA binding region fails p53 to activate transcription and act as co activator.
· Deletion in C-terminus prevents tetramerization and the protein is rendered inactive.
· Popova viral T antigen binds to the DNA binding region of the p53 and induces degradation and makes the cell p53 recessive.
· Over expression of mdm2 destroys p53.
· E1B (19kd) inactivates apoptotic activity of p53.
· E1B (55kd) blocks transactivation domain of p53, but it does not block its apoptotic activity.
· HPV E6 binds to p53 and targets for degradation.
· Adeno viral E1B binds to p53
· Loss of p53 leads to cell immortalization. This achieved by p53. Inactivation of Telomerase is normal in differentiated cells. Reactivation is necessary for reactivation of cells into cell division mode. Cells enter into crisis stage when telomeres become very short and cause genetic instability. TRF2 protect Telomeric ends, but when they are lost, 3’ends of Telomeric DNA are made available and p53 binds to DNA and gets activated. Now p53 rescues cells from crisis, blocks apoptosis, and lead to cell division.
Cell Cycle Inhibitors:
Inhibitors play an important role in controlling cell cycle events. Loss or mutation in them favors cancer development. Ink4A codes for transcript, which is spliced alternately to generate p19 and p16. p19 inhibits mdm2, so that p53 is saved from destruction. P16 inhibits Cdk4, 6-Cyclin kinase, so prevents phosphorylation of RB. Deletion of both is found in cancer cells.

In eukaryotic cells, there are multiple CDK-cyclin complexes that play specific roles at various phases in the cell cycle. These complexes include three interphase CDKs (CDK2, CDK4, and CDK6), a mitotic CDK1 (also known as cell division control protein 2 (CDC2)), and ten cyclins belonging to four different classes (A-, B-, D-, and E-type cyclins) (1). This specificity is described in Figure 14.2.3, where mitogenic signals correlate with the increasing expression of D-type cyclins. By binding with CDK4 and CDK6, activation of the kinases during G1 results in the initiation of the cell cycle where the cell prepares for DNA synthesis (1). Cyclin degradation is a crucial process that corresponds to the inactivation of CDKs, which in turn affects the progression of the cell cycle (Figure 14.2.3). According to the classical model of cell cycle control, cyclin D-CDK4 and cyclin D-CDK6 stimulate the initiation of G1 phase and the start of the cell cycle (not). The progression towards the end of G1 phase is characterized by the increasing levels of cyclin E-CDK2, which in turn triggers the onset of S phase. As discussed previously, the reduction in cyclin E-CDK2 levels by degradation is important in initiating the S phase. As seen in Figure 14.2.3, cyclin A-CDK2 regulates the completion of the S phase and entry into G2 phase, where cyclin A-CDK1 is involved. The level of cyclin B increases during the start of mitosis and diminishes at end of the M phase. The inactivation of CDK1 due to decreasing cyclin B triggers the end of the cell cycle. Overall, the life cycle of a cell is orchestrated based on the presence of various CDK-cyclin complexes, which can affect the progression of cellular proliferation depending on their presence and levels. This selectivity within the cell cycle offers potential targets for clinical therapuetics for cancers.
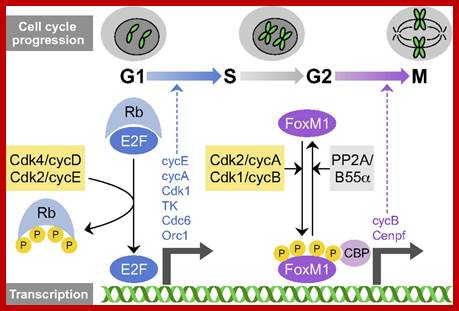
http://dev.biologists.org/
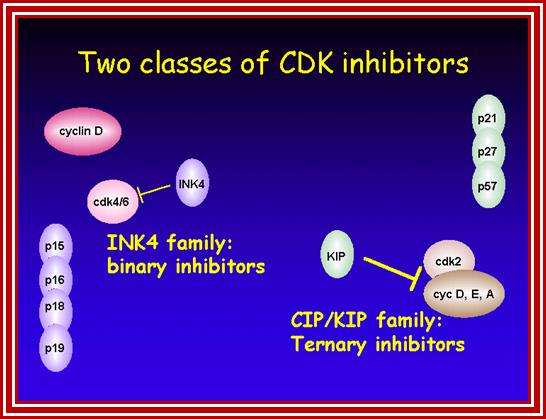
http://streaming.cineca.it/
P53 activation induces primary inhibitors such as p21 and p27. They block the cell entry from G0 to G1 and then block the entry into S phase. This critical regulation is achieved by inhibiting Cdk4, 6-Cyclin-D to block the entry from Go to G1. Then blocking Cdk2-Cyclin-E prevents the cell enter into S-phase. Such inhibitors effect is also on RB bound E2F transcription factors. If RBs remain unphosphorylated they sequester transcription factors, so cell cycle is blocked at G1 stage itself.
Cdk-Cyclin Inhibitors are grouped into two major classes;
1) INK4A family: They are made up of p16, p15, p18, 19; they are called binary inhibitors because they bind to kinase subunit and prevent the binding cyclin.
2) Cip/Kip: They are called ternary inhibitors, for they bind to Cdk-Cyclin complex; p21, p27, p57
Breast cancer:
Swelling, dry skin, shrunk and wrinkled breast skin, Lumps in the arm pit,
Lumps in the breast one can feel by themselves; lumps develop from epithelia
l cells of lactating tubules, nipples pulled in, one can find liquid discharges
from the nipples, breasts are swollen and reddish, Pregnancy at the age of
20-24 and 34-40, the later has greater probability of getting cancer, aged
women are more prone, women who does not feed their child with
breast milk, are prone to cancer; some time breast cancer and ovarian cancer
are common in the same women.
Ashkenazi Jews have greater propensity for breast cancer.
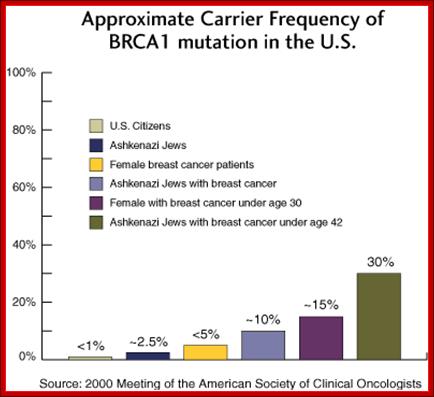
Global cancers; http://www.cdc.gov/cancer
![]()
https://plasticsurgeons.com.au/
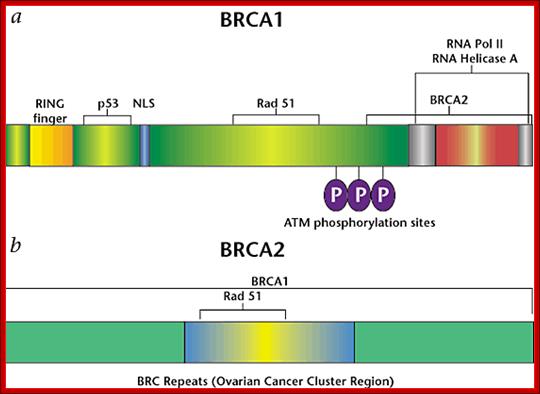
http://www.au-kbc.org/
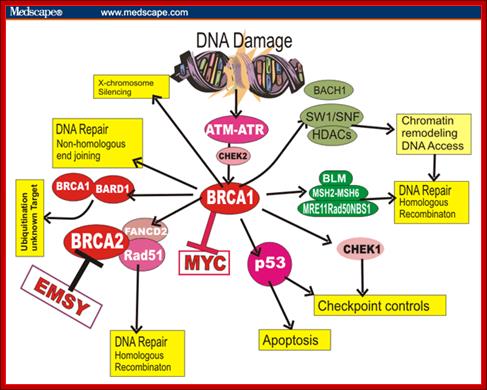
Examples of interaction involving BRCA1 and BRCA2 proteins, the exact order of interactions , evens and understanding of these are still evolving; http://www.medscape.com/
Most common cause of hereditary breast and/or ovarian cancer: Germline mutations in these genes are associated with an increased predisposition to breast and ovarian cancer and other types of cancer. More common in the Ashkenazi Jewish population. Genes/loci: BRCA1 (17q21) BRCA2 (13q12). Inheritance: Autosomal dominant.
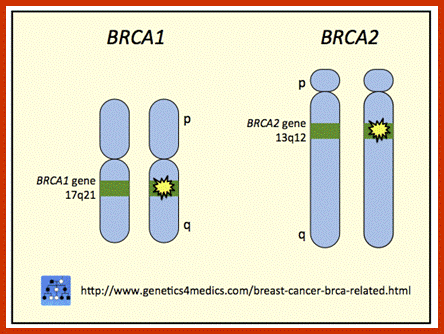
http://genetics4medics.com/
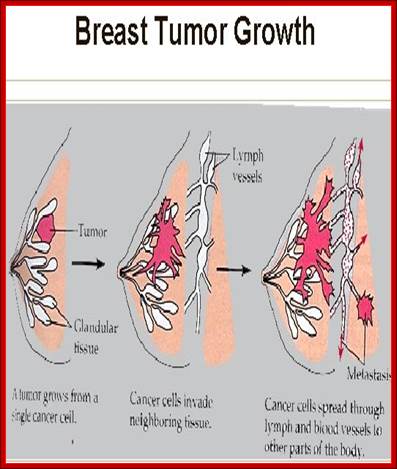
Genes:
BRCA1-Chromo-17
BRCA-2- chromo13,
D2 gene activates Brca2 while Brca1 is normal
Both are tumor suppressors, for they are responsible for DNA damage repair.
Estrogen receptors and progesterone receptors- can cause breast cancer with estrogen,
or hormone therapy.
BRAC-1 acts at G1-S check point. 30% of breast cancer cells over express Her2 gene,
this activates Cyt Oxidase II genes. Her’ is human epidermal growth factor receptor.
More than 50% of the genes are amplified in B/C,
More than 5 genes are over expressed in B/C.
Gleevac is used for breast cancer,
Herceptin-2 is used,
Blood Cancer:
In bone marrow stem cells differentiate and develop into different types of cells.
Some cells destined to be blood cells, some develop instead cancer cells, filling
the entire bone marrow causing under development of other cell types.
Acute leukemia- affect WBCs,
Chronic leukemia- myeloid leukemia.
Symptoms:
Anemia, Bruising,
Repeated infections,
Weight loss,
Night sweating,
Fever.
Chronic lymphocytic leukemia-
Enlargement of lymph glands in neck, groin and armpits, enlarged spleen.
Any gene abnormal for proliferation causes leukemia,
Colon Cancer:
Abdominal Pain,
Rough painful bowel movement accompanied with blood oozing,
Abdominal mass,
Constipation,
Stools narrow and thread like,
Weight loss,
Ever tired ness,
Vomiting,
Anemia,
Hundreds of polyps develop, polyp lesions, plate lesions, and loss of cell adhesion,
detected by colon endoscopy.
Genes- APC
Familial adenomatous polyposis FAP,
MSH2-and MSH6 (chromosome-6) DNA repair genes,
PIK3-p is mutated,
Mutation in RAS,
Chromosomal Instability,
Replication error,
Prostate cancer:
HPC1-Heriditory prostate cancer gene,
EphB2,
RNAse-L,
Chromosomal abnormality,
ETV 4 is over expressed,
Androgen gene receptor defect,
Symptoms:
Swelling of prostate, heavy urination at night, difficulty in urination,
inability to urinate, Painful-burning urination, discomfort, and erectile
dysfunction. And also painful ejaculation, blood in urine and semen,
frequent pain in the lower back of hips and upper thighs.
Advance Robotic Technique ART- prostatectomy.
Cancer Therapy:
Cancer therapy is wish among the doctors and patients. Cancer is due to multiple action of more than one gene or it can be one gene. If one identifies the gene that is responsible for a specific cancer, perhaps one day it can be cured. One method that has been in active research is immuno-therapy. If immune cells such as T cells can be triggered to attack certain cancer cell types based on their surface antigens, it sure cure. Perhaps the cure is not in near future, but certain drugs used prevent cell DNA replication, but it affects normal cells also.
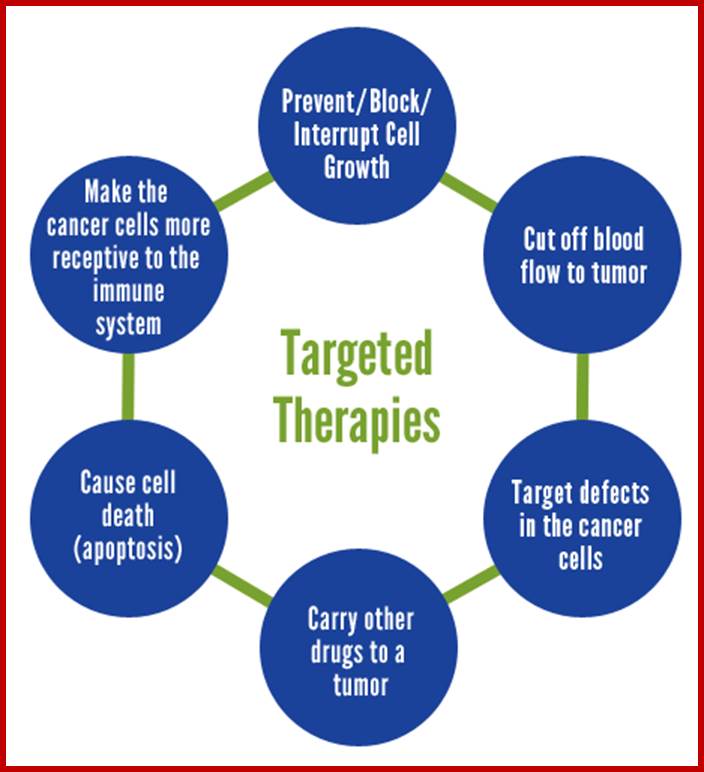 Preventing and fighting Cancer Naturally; http://www.livehealthynaturally.info/
Preventing and fighting Cancer Naturally; http://www.livehealthynaturally.info/
Brian J. Druker, MD;http://www.hhmi.org/research
Using an integrated functional genomics approach, we are currently cataloguing therapeutically targetable genetic drivers of leukemogenesis. We have developed two functional assays that identify tyrosine kinases essential for, and kinase inhibitors that affect, the growth of patients’ isolated leukemic cells. Using deep-sequencing methodologies, we then define the spectrum of mutations found within each patient’s cells. Next, we integrate the potential targets identified through our two assays with a patient’s mutational profile, which allows us to prioritize the functionally important mutations most likely driving the growth of that individual patient’s cancer. Each mutation is made and tested for its ability to promote growth factor–independent growth of a hematopoietic cell line model.
Using this methodology, we are beginning to annotate all the driver mutations found in less well defined leukemias and match them with potentially effective therapeutic inhibitors. For example, we have identified mutations in CSF3R, the receptor for granulocyte colony simulating factor (GCSF) in patients with chronic neutrophilic leukemia (CNL) and atypical CML (aCML). Through our functional assays and further characterization of these mutations, we have determined that, depending on the location of the mutation, CSF3R mutant leukemic cells respond to either ruxilitinib or dasatinib. The integration of the mutational spectrum with the functional data allowed us to define a novel subset of hematologic malignancies based on CSF3R mutational status and rapidly identify actionable targeted therapy interventions for these patients.
Using this integrated functional genomics approach, we hope to unravel the complexity of leukemia pathogenesis and provide the critical information necessary to discover life-saving drugs for patients with other types of leukemia.
 .
.
PNC-27 Nn Toxic Cancer Therapy; http://www.slideshare.net/

Potential cancer therapies inspired by mechanisms of extinction. FromCan oncology recapitulate paleontology? Lessons from species extinctions;
Viola Walther, Crispin T. Hiley, Darryl Shibata, Charles Swanton, Paul E. Turner & Carlo C. Maley; ;http://www.nature.com/nrclinonc/journal/;
Lessons from paleontology have taught us that one selective pressure is not enough to successfully eradicate cancer, but rather that multiple selective pressures are necessary, targeting the tumour by different mechanisms. The multiple pressures could target the different microenvironments in a spatially heterogeneous tumour. Paleontology concepts also suggest that we should apply combinations of chemotherapy, adaptive therapy, antiangiogenic therapy, hyperthermia, bacteriolytic therapy and targeted therapy over long periods, without breaks, to maintain selective pressures on the neoplastic cells, as is done with metronomic therapies.

MonoclonalAb.jpg; Monoclonal antibodies for cancer. ADEPT, antibody directed enzyme prodrug therapy; ADCC, antibody dependent cell-mediated cytotoxicity; CDC, complement dependent cytotoxicity; MAb, monoclonal antibody; scFv, single-chain Fv fragment. https://commons.wikimedia.org/wiki
Cell mediated Therapy:
Antibody-dependent cell-mediated cytotoxicity (ADCC);
Antibody-dependent cell-mediated cytotoxicity (ADCC) is a mechanism of attack by the immune system that requires antibodies to bind to target cell surfaces. Antibodies are formed of a binding region (Fab) and the Fc region that can be detected by immune cells via their Fc surface receptors. Fc receptors are found on many immune system cells, including natural killer cells. When natural killer cells encounter antibody-coated cells, the latter's Fc regions interact with their Fc receptors, leading to the release of perforin and granzyme B to cause tumor cell death (apoptosis). Effective antibodies include Rituximab, Ofatumumab, and Alemtuzumab. Third generation antibodies under development have altered Fc regions that have higher affinity for a specific type of Fc receptor, FcγRIIIA, which can dramatically increase ADCC..;
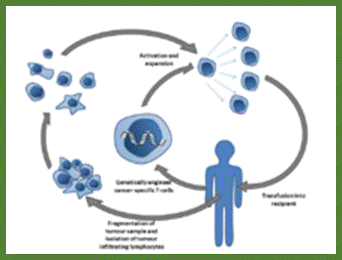
Adoptive T-cell therapy is a form of passive immunization by the transfusion of T-cells. They are found in blood and tissue and usually activate when they find foreign pathogens. Specifically they activate when the T-cell's surface receptors encounter other cells that display small parts of foreign proteins on their surface antigens. These can be either infected cells, or specialised immune cells known as antigen presenting cells(APCs). They are found in normal tissue and in tumor tissue, where they are known as tumor infiltrating lymphocytes (TILs). They are activated by the presence of APCs such as dendritic cells that present tumor antigens. Although these cells have the capability of attacking the tumor, the environment within the tumor is highly immunosuppressive, preventing immune-mediated tumour death; https://en.wikipedia.org/wik; https://en.wikipedia.org