Molecular Tools and Techniques-A
Transformation of Host Cells:
Introduction:
Once recombinant DNA is prepared, it is transferred into a host cell where it can be propagated, its gene can be expressed and the product is tested or recovered. The recombi8nant DNA can be used for a variety of purposes such as sequencing, promoter analysis, restriction site mapping, in vitro site directed mutagenesis and other genetic manipulations.
In order to have sufficient amount of the said Recombinant DNA, depending upon the functional and structural features of the R-DNA, one has use host cell that is suitable for the needs. Generally one expects that once the R-DNA is put into the cell, whatever may be the means, it should replicate in high copy numbers, it should not be restriction digested or modified; if the gene is expressed the protein should not be degraded or it should be secreted or localized as inclusion bodies, or it should be amenable to be isolated and purified, whether the recombinant DNA has really got the insert into the vector or not, or one should identify whether or not the cell has got the Recombinant-DNA. One also desires to have some inbuilt characteristic features, which are required for genetic manipulations. The host cells should also capable of taking in phage infection.
The bug, that yielded such variable and desirable features, is E.coli. Great advantage of E.coli is that it can be mutated easily and the mutants can be maintained. Even the bacterial genetic characters can be changed to the needs. Its cell cycle is just 30 minutes and one can grow them very fast and can get high yield. They can be subjected changed temperatures and nutritional conditions. Depending upon the needs one can use variants with specific characters.
Bacterial Cells used for transfection:
OD and cell numbers:
If E.coli cells are grown to 1 OD at 550nm, it is =10^8 cells.
If yeast cells are grown to 1OD at 600nm, it is = 10^7 cells.
Bacterial cells as host cells are used for transformation with simple plasmids, infection with lambda DNA, Pacmids and Bacmids. So for each kind of Recombinant DNA host cells have to have suitable or with compatible characters. Cells should produce 10^7 to 10^11 colonies per ug of plasmid DNA.
Mutants used:
|
Mutants |
Functional features
|
|
Rol-A
|
DNA multiplies to high copy numbers; no restriction is imposed for high copy numbers. |
|
Endo A (-) |
Endonuclease activity is absent |
|
Rec-A (-) |
No recombination (either site specific or homologous type) |
|
Tra (-) |
DNA is not transferred by conjugation |
|
Lon (-) d |
Cells have low protease activity against foreign EK proteins, |
|
Deo-R |
Cells can take large sized DNA |
|
Lac-I (Q) |
Cells over produce lac repressor, can be regulated |
|
Lac-z (d) M15 |
Contain lac-Z ‘w’ fragment, when induced produces ‘w’ fragment and complements with alpha fragment produced by the plasmid |
|
F’ |
Contain F1 plasmid produce sex pili required for infection with male cells (M13 phages) |
|
Sec (+) |
Secrete proteins efficiently |
|
Lambda inf (+) |
Compatible for lambda infection |
|
PI-inf (+) |
Compatible for infection with PI phages |
|
Thi (-) |
Requires thiamine for the growth of cells |
|
Rec-A/rec-B |
Lack recombination and the inserts are stable |
|
Hfla-150 (-/+ |
(-) Lack high frequency lysogeny- it means lytic; (+) high frequency lysogeny |
|
Lac-IR (+) |
Contain cI repressor activity |
|
hsd R17(17) |
Restriction minus. Modification positive, Foreign DNA not cleaved restriction endogenous enzymes |
|
Rk (-) |
|
|
Mk (+) |
|
|
rpoH |
Cloned proteins are stable. Doesn’t produce heat stress induced proteases |
|
McrA (-), McrB (-) mcrR(-) |
Blocks restriction of C methylated sites Lack restriction at methylated C in G*C Lack restriction at methylated C in G*C |
|
mrr(-) |
Lack restriction of methylated sites at C*AG and G*AC |
|
hsd-S(rB-mB) |
Restriction minus and methylation minus |
|
SupE/F |
Suppressor mutation, amber (UAA) and ochre (UGA), respectively at low Tm |
|
Ung (-) |
Lack uracil glycosylase |
|
Hsd S |
Restriction minus, modification minus, DNA is stable, but protective methylation is lacking |
|
Rel A |
Allows RNA synthesis in the absence of protein synthesis |
|
Tra-D |
Prevents transfer of F’ episome |
|
Delta-lon |
Mutation in protease, increases the stability of fusion proteins |
A list of bacterial strains used:
Bacterial strains such as JM109, DH5alpha and NM522 have most the above said features. Theses are the work horses for basic molecular cloning type of work.
Y1089: F’, delta lacU169, proA+,
Delta-lon, strA+, hflA150 (increased lysogeny), Tn10 (Transposon encodes Tetracycline resistance gene), can be induced by IPTG.
Y1090: similar to the above, but it can be screened by blue And white colony screening protocols. It is lytic, has Pmc (AMP^+ and Tet^ + helper Phage?
The helper phage is used super infect certain bacterial strains containing F’ or M13 phage Origin segments. This helps in producing single stranded DNA (either + or- strands), and such DNAs are very helpful in sequencing reactions.
Am-1 Cre strain: This strain produces recombinase enzyme called ‘Cre’. It brings about recombination between Lox sites found in plasmids like lambda DR2.
Y 1089 (Lysogenic): used for lambda GEM 11 and 12 vectors
Y 1090 (Lytic): used for lambda derived EMBL 3 and 4 vectors.
ED 8767: this is used for cosmid vectors like PFLR and Lawrist-4 vectors.
NS 3622: These are used for ‘Pacmid’ vectors such as pAd-10. And the host has repressor gene, which represses transcription when grown in the absence of sucrose.
NS 3529: These are used for Sac-B1 type of vectors. Cells have Cre^ +, RecA^-, lac-Iq, MCR ABC, and mrr.
JM109 (stbl-deor): for Bacmids such as pBeloBac-II:
JM 109 DE3: It is required for pET expression vector for it contains T7 RNA-pol gene promoter. The host contains T7 RNA polymerase gene under the control of lac-Z promoter operator and lac-Iq which produces repressor for lac Z promoter –operator. By adding IPTG the T7 RNA-pol gene is expressed and the RNA-pol transcribes the DNA that is under the control of T7 RNAP promoter. This host can also be used for the expression of pTac vector for it contains promoter and lac-Z promoter operator required for the expression.
Host cells used for Certain Vectors:
|
Vector |
Host strains |
|
Lambda GEM 11 and 12 |
Y1089 lysogenic Y1090 lytic |
|
Cosmid – pFLR |
ED8767 |
|
Lawrist-4 |
ED8767 |
|
Pacmid-pAd10/Sac BII |
NS3622, NS 3529 |
|
Bacmid |
|
|
PET and pTac |
JM109 DE3 or BL21 (De3) |
Lambda GEM 11 and 12, EMBL 3 and 4- host strains are Y1089 lysogenic, Y1090 lytic methods.
Most commonly used bacterial cells are:
JM 109,
JM 109 (STBL2),
DH 5 alpha,
DH 10 B,
NM 522,
DH 5 alpha mcr
7-B. Host cell Transformation:
Bacterial cell transformation:
In order to transform bacterial cells, one has to select proper or required bacterial strains and prepare them for transforming with foreign DNA. The cells required for transformation are called competent cells.
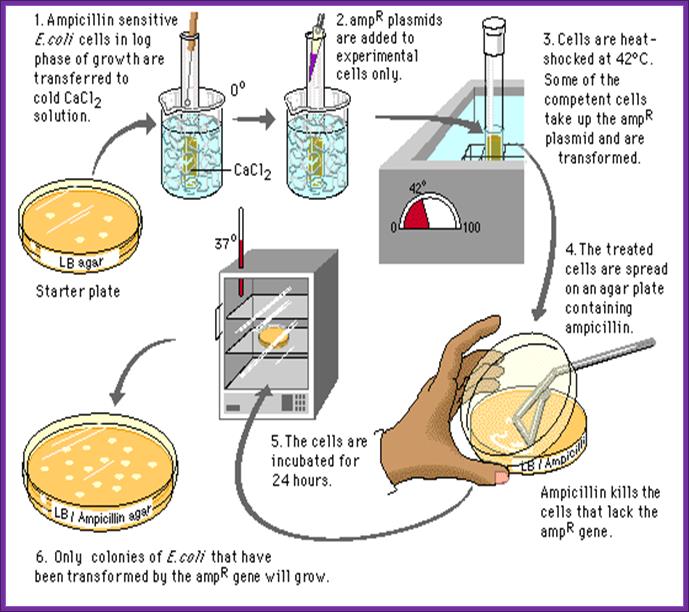
https://staff.rockwood.k12.mo.us;
Preparation of Competent Cells:
- Pick fresh colonies from the agar plate and suspend the same in 1 ml of SOB.
SOB: Bacto Tryptone 2%,
Bacto yeast extracts 0.5%,
NaCl 10mM,
KCl 2.5mM.
Growth medium:
SOB pus MgCl2 and MgSO4 22mM each,
(Add 10 ml each from 2M stock solution).
- Inoculate cells into 50ml of growth medium in a 250ml of conical flask and grow them on an incubator shaker overnight.
- When cells reach an OD of 0.5 or 0.7 at 550nm (that is + 4-7x10^7 cell/ml), harvest the cells then keep them on ice for 15-20 minutes.
- Suspend the cells in 1/3 volume of the original quantity of growth medium, in TFB. Gently shake till all the cells are freely suspended.
TFB:
K-MES 10mM (2-(N-morpholino) ethanesulfonic acid)
KCl 100mM,
MnCl2 2H2O 45mM,
CaCl2 2H2o 10mM,
HACOCl3 3mM, (hexamminecobalt trichloride).
Adjust the pH to 6.2.
FSB:
K.acetate 10mM,
Redistilled Glycerol 10%,
KCl 100mM,
MnCl2. 2H2O 45mM,
CaCl2. 2H2O 10mM,
HACOCl2 3mM. pH 6.2.
- Incubate cells in the said medium for another 15 minutes on ice. Pellet the cells 4000 rpm for 5 minutes. All the procedures should be done at 4^oC.
- Resuspend the cells in 1/12.5 volume of the TFB.
- Then aliquot 200ul of them to each tube.
- Add7ul of DMSO (Dimethyl sulfoxide) and DTT (Dithiothreitol).
to 3.5% (V/V) to each tube.
Now the cells can be used for transformation. Such cells are called competitive cells or competent cells. During treatment with CaCl2 or Rubidium chloride and Manganese chloride, cell walls are relaxed and the proteins required for the uptake of DNA get localized in the plasma membrane. If we want to store these cells at -70^oC for some period of time it can be done provided the cells are suspended in 1/3 volume of FSB buffer and then pellet and resuspend in the same FSB buffer in the same amount. After final pelleting they are added with DMSO to 3.5%, no DTT. Keep the cells on ice. Then aliquot cells into tubes and it can be stored at -70 for at least 2 months.
- Freshly prepared cells or thawed competent cells can be used for transformation.
- Keep the cells on ice.
- For 200 ul of cells (10^7 cell per 200 ul) add 1 ul of purified ligated mix, in such a way there should be one DNA molecule for every ten or more bacterial cells.
- Keep them on ice for about 20-30 minutes.
- Transfer the tube to hot water bath for one to two minutes at the temperature of 42^oC. This ‘heat shock’ is believed to increase the efficiency of transformation. How?
- Cool the cells on ice.
- Add 500ul of recovery medium and incubate for one hr on an orbital shaker at 37^oC.
Recovery medium:
SOB (-Mg),
MgCl2+MgSo4 to 22mM,
Glucose 2%
- Then pour the same on to agar plate containing 100ug/ml of Ampicillin. Ampicillin should be from fresh stock and always check its viability for the AMP gets degrade at room temperature and it cannot be stored for a long time.
- Transformation efficiency has been found to be 10^8 to 10^12 per ug of pUC plasmids.
- Efficiency transformation decreases as the size of the plasmid increase from 3000 bp to 10 kb or more. Only circular DNA is capable transformation but not linear DNA. DNA, which is free from protein, is more efficient in transformation than the DNA contaminated with proteins.
During intake of DNA certain proteins found in the membranes play an important role. When cells are grown in optimal conditions and treated with MnCl2 or Rubidium chloride these proteins are synthesized. They some how induce the cells into competency.
When ds DNA comes in contact with membrane bound proteins, they hold the DNA and produce a nick in one of the strands and the 3’ end of the nicked strand is pulled into the cell and the same is converted into double stranded circular structure. The exact details are still speculative.
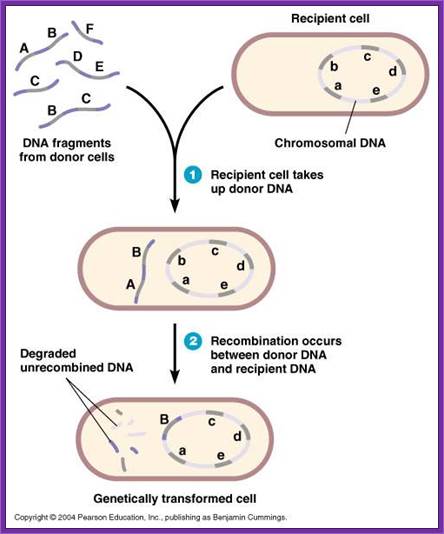
Note- difference between exogenote, endogenote and merogenote; http://www.biologyexams4u.com/
DNA transferred from outside is called exogenote, the native DNA inside the cell is called endogenote; and resultant partial diploid is called merozygotes. Any process of genetic exchange that transfers only a part of genetic material from one cell to the other is called meromixis.
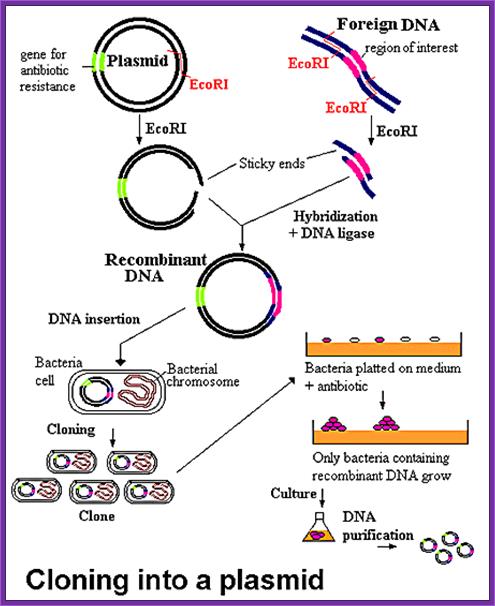
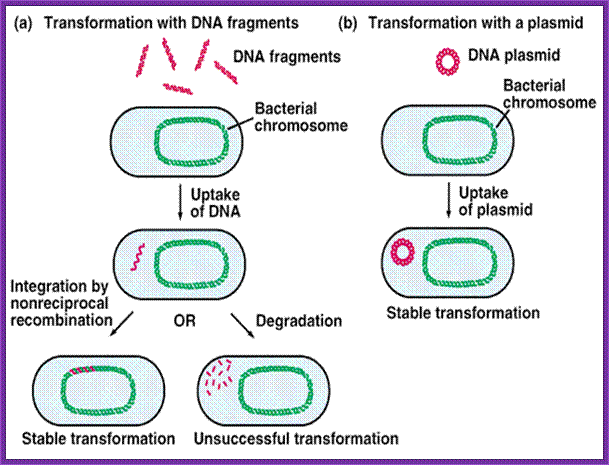
http://www.biologyexams4u.com/; http://mybedsidemanner.blogspot.com/
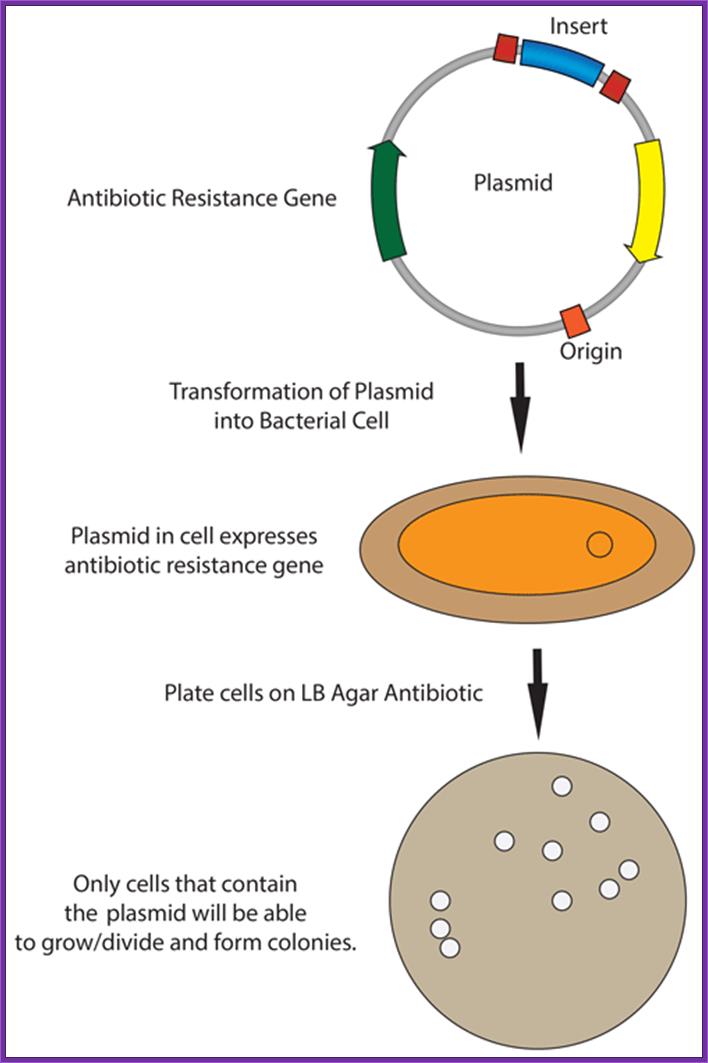
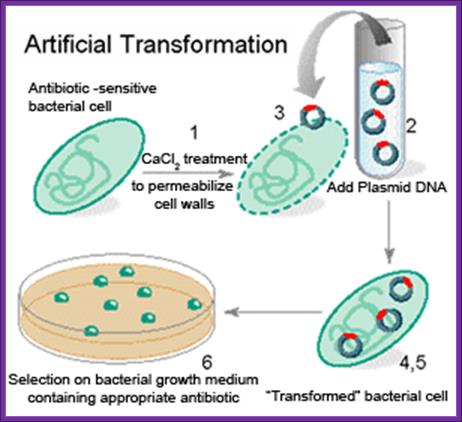
- Cells must be made competent; 2 plasmid DNA must remain intact before it is introduced; 3. Dna must be translocated across the cell wall; 4. DNA must not be degraded by restriction enzymes in cytoplasm, 5. DNA must expressed and should be monitored whether or not it is expressed (use a marker); http://www.odec.ca/; image from Cohen/Cohen_4.html.
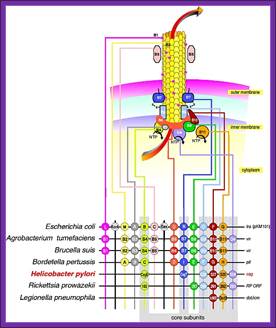
The figure shows the cellular components involved in transport of their plasmid DNA to the acceptor cells. Uptake of DNA requires proteins that are related to those involved in the assembly of type IV pili and type II secretion systems, as well as a DNA translocase complex at the cytoplasmic membrane; DNA uptake during bacterial transformation /DNA transportation across the cell wall; http://pathogenomics.bham.ac.uk/
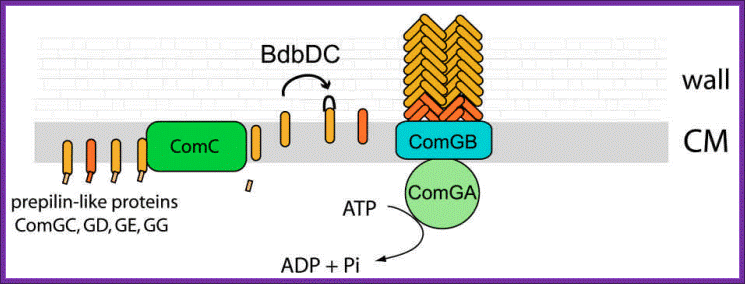
Figure1. DNA transport during transformation; Model for the biogenesis of the DNA binding/transport machinery in Bacillus subtilis. Prepilin like proteins ComGC, GD, GE and GG are processed by prepilin peptidase ComC, which cleaves the short leader peptides at their N-terminus. The mature ComGC is ranslocated across the membrane, and its two cysteine residues are oxidized by BdbDC, forming an intramolecular disulfide bridge. ComGC forms a polymeric complex which traverses the cell wall and allows the DNA-binding protein ComEA to access exogenous DNA. ComGA a). traffic NTPase), ComGB (a polytopic membrane protein) and the other pilin-like proteins are necessary for the complex formation, and it is possible that the minor pilinlike proteins may have structural roles. The major pilin-like protein (ComGC) is represented in orange and the minor pilin-like proteins in red. Ines Chen And David Dubnauhttp://www.frontbiosci.org/
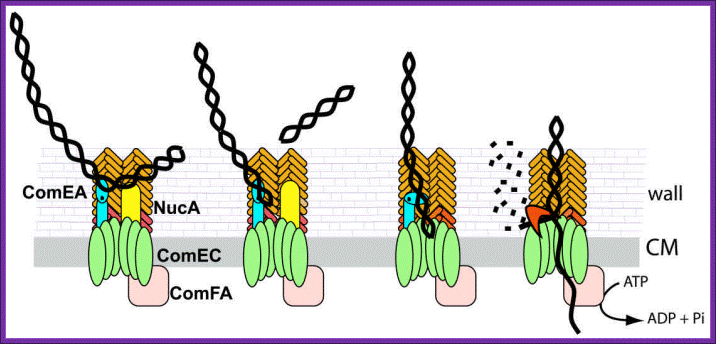
Figure 2. Model for DNA uptake in B. subtilis. Exogenous DNA binds to its receptor, ComEA, and perhaps to other protein(s) on the bacterial surface. The DNA undergoes endonucleolytic cleavage by NucA, leading to formation of new free termini. ComEA undergoes a conformational change, perhaps by bending at its hinge region, and delivers the DNA molecule to the channel formed by ComEC, which traverses the cytoplasmic membrane. The translocation of the DNA molecule to the cytosol as a single-stranded molecule is catalyzed by ComFA. The nontransported strand is degraded to acid soluble products by an unidentified nuclease. The major and minor pilin-like proteins are color coded as in Figure 1. http://www.frontbiosci.org/
- Degradation of incoming DNA and transformation defect in S. pneumoniae dprA and recA mutant cells: Mathieu Bergé,
- Isabelle Mortier-Barrière, Bernard Martin, Jean-Pierre Claverys
Our observations establish for the first time that transforming DNA requires protection from endogenous DNase(s) immediately after uptake by S. pneumoniae competent cells. Whereas in wild-type cells about 25% of incoming ssDNA is processed into recombinants (Lacks, 1962; Vijayakumar and Morrison, 1983; Méjean and Claverys, 1984), in absence of either DprA or RecA resulted in incoming ssDNA being very rapidly degraded. The nuclease responsible has not yet been identified, but could be a 3′→ 5′ exonuclease, as donor strands enter 3′ first (Méjean and Claverys, 1988; 1993). It has early access to incoming ssDNA and so is likely to be located close to or be part of the entry pore. It could be related to the nucleolytic activity recently proposed to be associated with the C-terminal domain of ComEC (Bergéet al., 2002), a protein believed to form an aqueous pore for DNA uptake. Thus, DprA could be primarily responsible for protection of incoming ssDNA. It could perform this role in two different ways, by interacting directly (i) with the putative DNase to prevent its access to incoming ssDNA, thus permitting the loading of RecA, or (ii) with the 3′ end of incoming ssDNA, to cap it and thus protect it from the putative DNase. However, neither hypothesis explains why inactivation of recA provokes destablilization of ssDNA to the same extent as the dprA mutation. A possibility would be that RecA is required to activate DprA, for example via its coprotease activity (Steffen and Bryant, 2000).
DNA transport into Bacillus subtilis requires proton motive force to generate large molecular forces; Berenike Maier1, 3, Ines Chen2, David Dubnau2 & Michael P Sheetz1
Bacteria can acquire
genetic diversity, including antibiotic resistance and virulence traits, by
horizontal gene transfer. In particular, many bacteria are naturally competent
for uptake of naked DNA from the environment in a process called
transformation. Here, we used optical tweezers to demonstrate that the DNA
transport machinery in Bacillus subtilis is a force-generating motor.
Single DNA molecules were processively transported in a linear fashion without
observable pausing events. Uncouplers inhibited DNA uptake immediately,
suggesting that the transmembrane proton motive force is needed for DNA
translocation. We found an uptake rate of 80 ![]() 10 bp s-1 that was force-independent at external
forces <40 pN, indicating that a powerful molecular machine supports DNA
transport.
10 bp s-1 that was force-independent at external
forces <40 pN, indicating that a powerful molecular machine supports DNA
transport.
Natural competence (from Wikipedia):
Main article: Competence (biology).
About 1% of bacterial species are capable of naturally taking up DNA under laboratory conditions; more may be able to take it up in their natural environments. DNA material can be transferred between different strains of bacteria, in a process that is called horizontal gene transfer. Some species upon cell death release their DNA to be taken up by other cells, however transformation works best with DNA from closely related species. These naturally competent bacteria carry sets of genes that provide the protein machinery to bring DNA across the cell membrane(s). The transport of the exogeneous DNA into the cells may require proteins that are involved in the assembly of type IV pili and type II secretion system, as well as DNA translocase complex at the cytoplasmic membrane.
Due to the differences in structure of the cell envelope between Gram-positive and Gram-negative bacteria, there are some differences in the mechanisms of DNA uptake in these cells; however most of them share common features that involve related proteins. The DNA first binds to the surface of the competent cells on a DNA receptor, and passes through the cytoplasmic membrane via DNA translocase. Only single-stranded DNA may pass through, one strand is therefore degraded by nucleases in the process, and the translocated single-stranded DNA may then be integrated into the bacterial chromosomes by a RecA-dependent process. In Gram-negative cells, due to the presence of an extra membrane, the DNA requires the presence of a channel formed by secretins on the outer membrane. Pilin may be required for competence however its role is uncertain. The uptake of DNA is generally non-sequence specific, although in some species the presence of specific DNA uptake sequences may facilitate efficient DNA uptake.
http://neuron.mefst.hr/

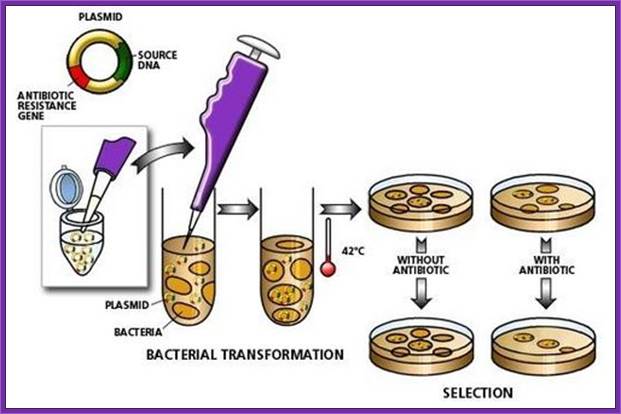
The recombinant plasmids and the bacterial cells are mixed up. Plasmids enters the bacteria in a process called transfection. With the recombinant DNA molecule successfully inserted into the bacterial host, another property of plasmids can be exploited - their capacity to replicate. Once inside a bacterium, the plasmid containing the human cDNA can multiply to yield several dozen copies.When the bacteria divide, the plasmids are divided between the two daughter cells and the plasmids continue to reproduce. With cells dividing rapidly (every 20 minutes), a bacterium containing human cDNA (encoding for insulin, for example) will shortly produce many millions of similar cells (clones) containing the same human gene.; BIO. "Biology in Perspective." Washington, D.C.: Biotechnology Industry Organization, 1990.; http://www.accessexcellence.org/
Competent cells for Electroporation:
· Grow cells from a single colony in SOB overnight. Then transfer 7.5ml of this to750ml of SOB and grow till the density reaches .5 to 1 OD at 550nm.
· Collect the cells by centrifugation.
· Suspend the cells in 10% glycerol in ultra pure water. Keep the cells on Ice for 15 minutes.
· Spin to collect the cells and suspend in 10%glycerol. Keep the flask on ice.
· Then spin the cells to collect the cell and suspend the cells in the same residual liquid.
· Aliquot the cells in 200ul each containing 10^7 cells per vial.
· Such cells can be frozen and stored at -70^oC.
Electroporation:
- Take 20 of such cells into .15cm cuvette or 40ul in .2cm cuvette. Add1 ul of ligated recombinant DNA suspended in TE. To remove any conductive ions by floating the ligation liquid on Millipore filter type .00um pore size for 3-4 hrs. Then recover the liquid and use the same.
- As far as possible avoid or reduce metal ion concentration.
- Perform electrophoresis at 12.5Kv/cm^2, at 25mF capacitance and 200 ohms resistance for 5 milliseconds.


|
Order No. |
Description |
Qty |
|
EC1 |
1 mm gap cuvette |
50 |
|
EC1L |
1mm gap cuvette (long electrode) |
50 |
|
EC2 |
2 mm gap cuvette |
50 |
|
EC2L |
2 mm gap cuvette (long electrode) |
50 |
|
EC4 |
4 mm gap cuvette |
50 |

- Remove the cells and keep the same on ice and add recovery medium and incubate for 1hr on incubator shaker at 37^oC and plate the same on nutrient Agar plate containing 100ig/ml of Ampicillin and spread them uniformly.
- During electroporation the current applied creates a kind of porations in the membrane and also drives the DNA into the cells across the membrane. Efficiency of electroporation is very high provided the cells are prepared properly. This can be any thing up to 10^10 to 10^12 per ug of DNA.
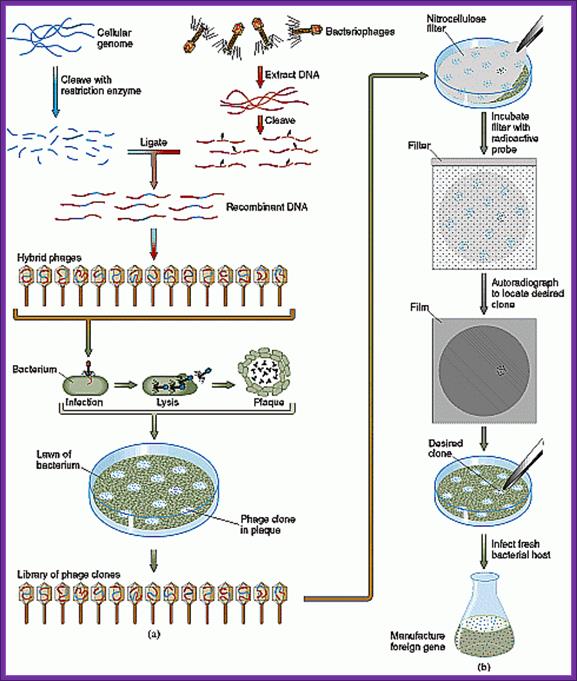
Screening of Plasmid based cDNA library:
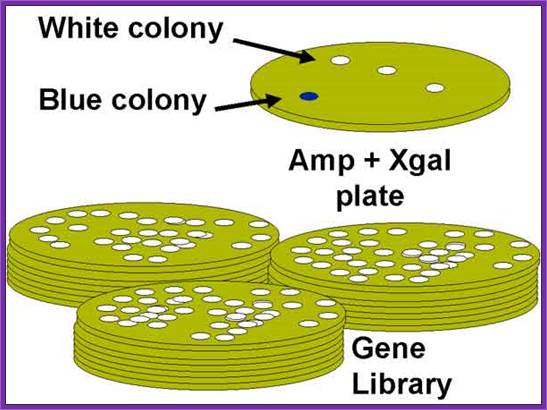
Once bacterial cells are transformed with recombinant DNA, they are spread onto agar plate containing nutrient medium plus 100ug/ml Ampicillin and IPTG and X-Gal. IPTG induces the expression of the DNA that is inserted next to the Lac-Z promoter. X-gal acts as the substrate for beta Galactosidase the functional form, which is produced when the alpha subunit from the plasmid and omega subunit are expressed by the host cell. If the functional enzyme is produced it acts on X-gal cleaves the glycosidic bond and produces blue color. Those colonies that produce blue color are the ones that don’t have any cloned material. Those colonies that don’t produce any color are called white colonies. The white colonies represent where the insert i.e. recombinant DNA is inserted into cloning site, thus disrupts the production functional alpha peptide.
All those colonies that are white are the colonies that contain the foreign gene or foreign DNA. Whether the inserts are functional or not it is difficult to say at this point of time.
Library screening:
The cDNA represent the entire population of mRNA produced in that tissue. Depending upon the tissue the number of mRNA species range from 17000 (liver) to 22000 (Brain); which also means that these are the number of genes expressed. The copy number of each species may vary from 7-8 to 50000 or more. Preparation of cDNA has virtually converted mRNA to their respective genes. If the cDNA preparation is good all of them have potentiality to express the protein products. However translational initiation in prokaryotic requires shine Delgarno sequence in the leader region of mRNA, but the eukaryotic mRNAs contain Kozak sequence for initiation. Yet there is possibility some of the mRNA expressed in the transformed cells may be translated.
http://passel.unl.edu/
When once get a whole number of colonies, in order to get full representation of the whole library at least one has to screen 100, 000 colonies. In order to screen one should have probes for each of the genes.
On the other hand if one has probes for one or two genes either in the form of antibodies or oligomers one can screen the colonies to the clones.
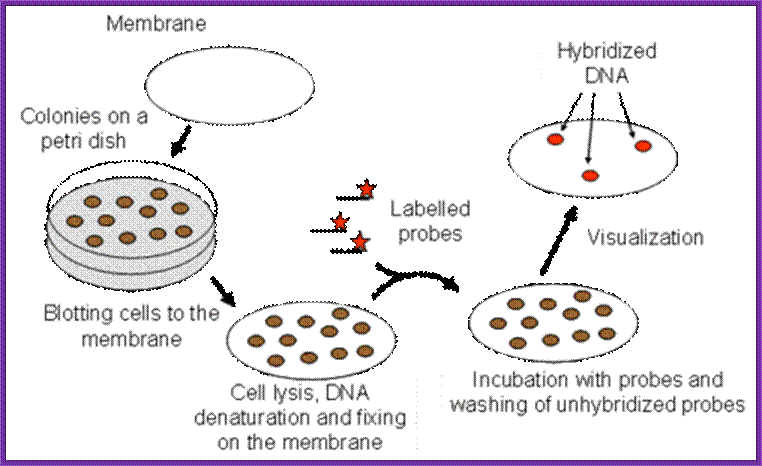
Screening for expressed protein products:
- This is more or less similar to that of western blotting.
- Take out the plates; mark them with numbers and also position of the colonies in the plate.
- Prepare nitrocellulose membranes soak them in buffer and place the membranes on the colonies without disturbing the colonies.
- Mark the positions of the membranes with respect to the plate. Keep the membranes for some time 15-30 minutes.
- Then remove the membranes carefully and place them on the freshly prepared nutrient gar plates in such a way the colonies adhered to the surface of the membrane are on the upper surface of the membrane.
- Grow cells over night in an incubator till they grow to pin head size.
- Then the membrane is treated with cell lyses buffer so that cells are lysed and contents spill over the cells. Carefully wash the membranes in PBS or TBS buffers, PBS+ 10mM Na phosphate pH 7.0, 150mM NaCl and add 0.1% Tween to make it as PBST. TBS consists of 50mM Tris-Cl, pH 8.0, 150mM NaCl.
- Then treat the membranes in PBS buffer containing skimmed milk (0.5% to 3%), which acts as the blocking reagent.
- Then place the membranes in PBS or TBS and add antibodies.
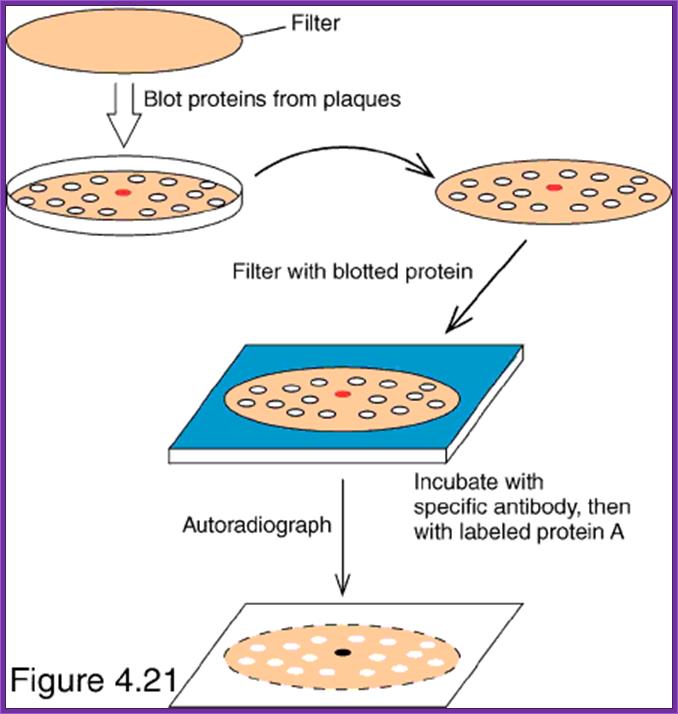
- Allow antibodies to bind to their respective colonies where the desired protein is expressed.
- Then wash the membranes in proper buffers and add anti-antibodies conjugated with alkaline Phosphotase. Then wash the membranes to remove any free anti-antibodies.
- Then add alkaline Phosphotase buffer and Nitro blue Tetrazolium (100 ug/ml) and BCIP (50 ug/ml). Keep it in dark for few minutes. Wherever the desired protein is produced it produces purple color, which is a positive indication that a colony has the desired gene.
- By proper positioning of the Nitrocellulose membrane having the said colony one can pick up the colony from the plate and the same can be tested for the protein and then it can be used for a variety of purposes.
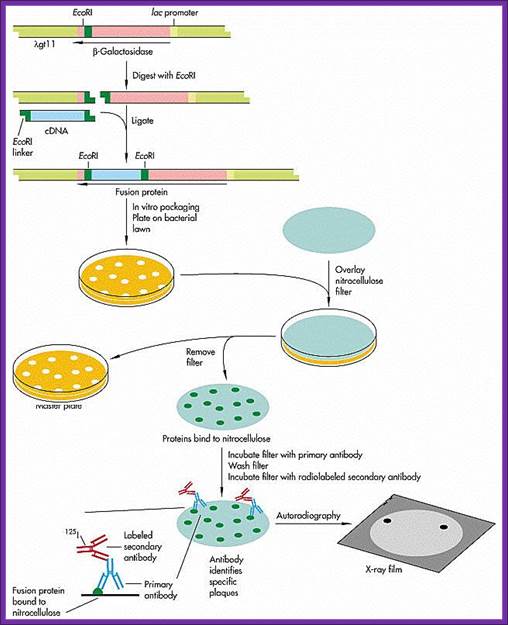
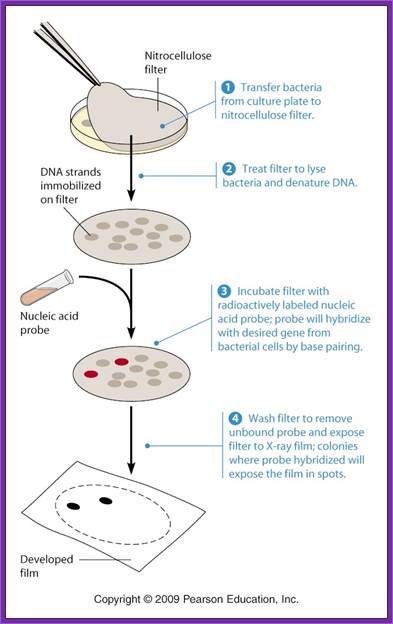
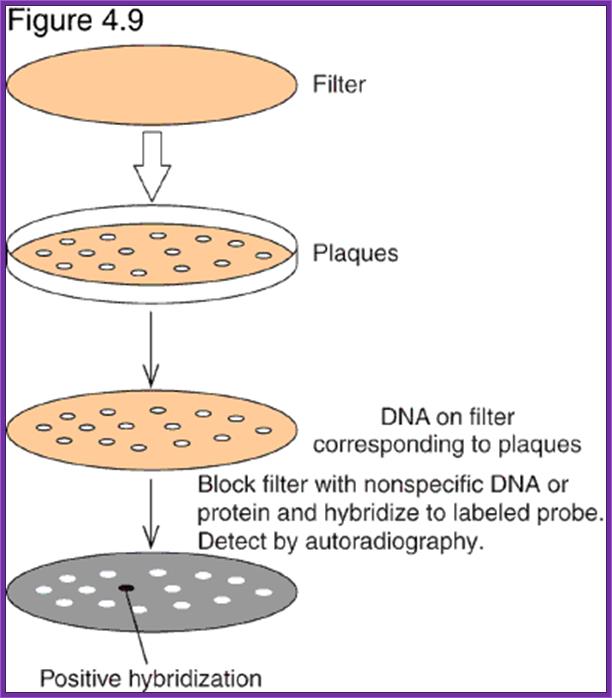
Screening the library with DNA Oligomer probes:
The protocol is more or less similar to that of southern blot technique. In this protocol one can use a known DNA segment or known RNA as probes. They should be labeled either by radioactive isotopes and nonradioactive labeling compounds.
The ratio between blue-white colonies indicates how many colonies contain cDNA inserts.
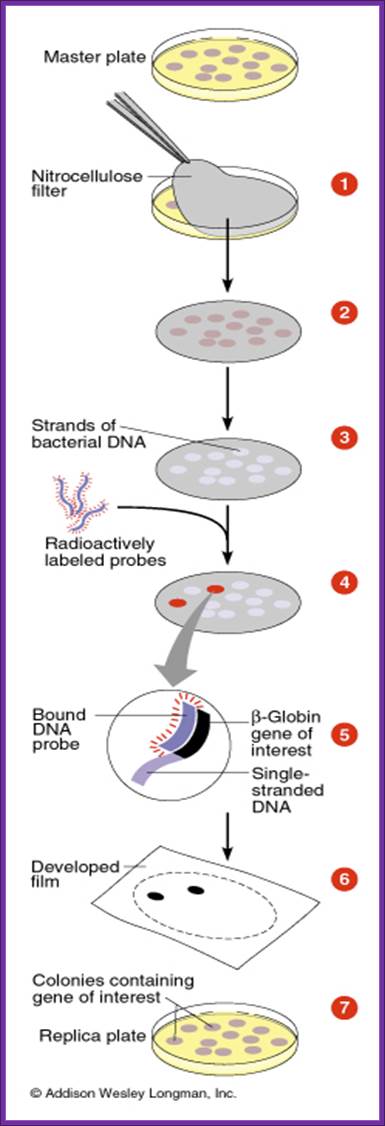

- The colony containing plates are to be numbered and marked for the position of colonies.
- Nylon or Hybrid membranes or Nitrocellulose membranes of suitable sizes are soaked in Hybridization buffer or SSc buffer.
- The membranes are laid on the agar plates containing colonies gently and without disturbing colonies.
- Leave the membrane for 30 minutes. Mark positions on the membrane in relation to the position of colonies in the plate.
- Remove the membranes and soak the same in cell lysing buffer and then it is treated with DNA denaturing buffer such as 0.4 M NaOH for 3-4 minutes, then the same is neutralized.
- Such membranes are exposed to ultraviolet rays for fixing DNA backbone to bind to the membranes. Other wise the membranes are backed in vacuum-hot air oven for 90 minutes at 95^oC. This fixes single strand DNA to the membranes.
- The membranes are soaked in Hybridization buffer and they are incubated with pre-hybridization buffer containing calf thymus sheared DNA fragments for about 2-3 hours.
- Then the buffer is decanted and the probe, it can be labeled DNA or labeled RNA, is added to the plated with hybridization buffer and incubated at 55^oC in a hot water shaker water bath. Generally the membrane and the probe are added into polythene bags and they are sealed in such a way all air bubbles are removed.
.
- After 36 to 48 hrs, the probe is decanted and preserved for another experiment; for it can be used for few more experiment with out loosing much of its activity.
- The membranes are washed in buffers and air dried and marked with ball point pens and exposed to X-Sheet if the label is radioactive isotope or if the label is luminescent, it can also be exposed to x-ray films, or the label is chromogenic it can be photographed.
- If one is looking for RNA as an expression product, one can follow the same protocol and probe with labeled DNA.

Lambda DNA based cDNA library:
There are a variety of designed lambda DNA vectors, which can be used for preparing cDNA library or genomic library. Once the cDNA or genomic DNA fragments are ligated vector lambda DNA vectors, they are to be extracted with phenol: chloroform. Then the ligated DNA has to be packed into fully formed phages.
The lambda DNA is about 48.5kbp long, it is ds DNA with single stranded tails of 12 ntds long; they are linearly organized when they are packed into the phage head, but when they are injected into the bacterial cell they circularized by base pairing with the ss tails which are complementary to each other. Hence this region of complementary sequences responsible for circularization of the linear DNA is called COS site (complementary sequences).
http://en.wikipedia.org/
An enzyme called Terminase, which is the product of one of the lambda genes, recognizes this sequence and it cuts to produce single stranded tails. At the left end of the Cos site about 200 ntds inwards a sequence is present and it essential for packaging the DNA into the prohead of the phage. For that matter all viral genomes contain packaging sequence for packing the genome into their capsid cages.
------------------- 5’GGGCGGCGACCT------------
-----------------------CCCGCCGCTGGA5’ ---------
In order to pack the recombinant DNA into phage heads one has to obtain phage packaging extracts. These are specially prepared from two different mutant strains. At the time when phage capsid, tail, tail fibers and other proteins are produced in a specific bacterial strain the bacterial cells are lysed and lambda DNA and other components are digested.
The extract prepared contains all the components required for producing functional phage particles when the required phage DNA is provided. The phage vector DNA is approximately 43.5kbp. On one side of the DNA it has sequences required for packing the DNA into the prohead of the virus. If a foreign DNA either in the form of cDNA or genomic DNA is ligated into such vector DNA which has 12 ntds long tails with complementary sequences called COS site and packaging signals. When such DNA is added into phage packaging extracts, the lambda DNA is recognized by its sequence at the Cos site and the same is threaded or loaded into the prohead till the other end with Cos site reaches the orifice at the entry of the head. At this point if there are any concatameric DNA, it is cut at the site and one full-length phage DNA is loaded. Similarly other DNA lengths are loaded into preformed prohead. Once the DNA is packed into the head other structural elements such as collar, tail tube, tail fibers and tail plate join. And tail fibers are added late to form a functional lambda phage. The number of phage particles assembled depends upon the quantity of the packaging components in the extract and the number of DNA fragments available at the time of packaging.
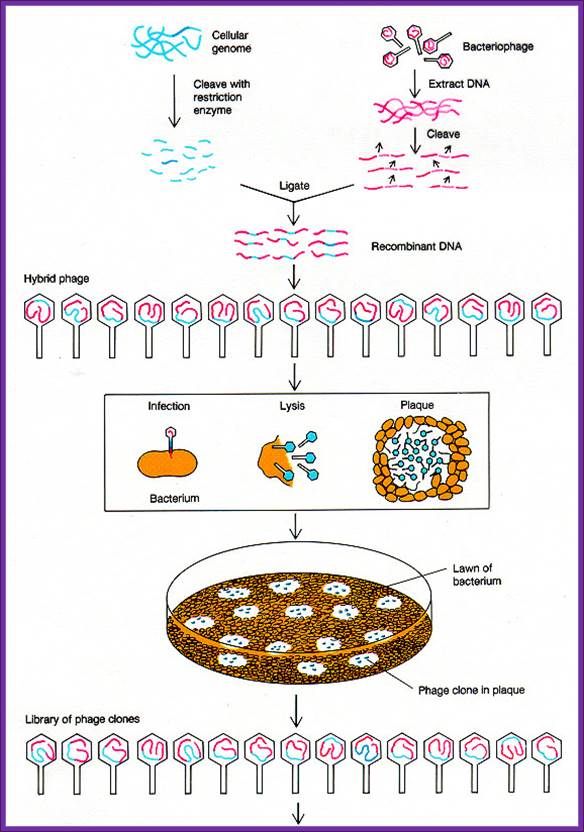
Packaging extract preparation requires expertise and experience and it is time consuming. Many companies make such extract and sell the same. Most of the extracts are aliquoted (50ul per tube) and shipped in dry ice. These have to be maintained at -70^oC. Once the extract is thawed they cannot be reused. Most of the packaging extracts of 50ul per tube can generate 10 ^7 to 10^8 particles.
Viral Packaging:
Thaw the required amount of packaging extracts on ice. The dilute the recombinant DNA to the required concentration. Then add 5ul of the DNA (approximately 0.5 to 1 ug of DNA) is added to every 50ul of the extract, which can easily produce 10^10 to 12 phages (theoretically) and gently tap the tube and keep it at room temperature for two hours.
Then add phage buffer 500 ul and add 25ul of chloroform and mix it gently by inverting the tubes. Chloroform clarifies the mix and inactivates all other components of the extract that is not used. Phage particles are not damaged. In this state the phage particles can be stored at 4^oC for 3 weeks and at -20^oC for a month or more. For long term storage add S-buffer extract with chloroform and take out the supernatant add DMSO to the final concentration of 7%. The same can be aliquoted and stored at -70^oC for six to 10 months.
After two hrs and chloroform extraction the packaged viruses are diluted 1:1000 or 1:10000. This type of dilution is required for optimizing pfu per ml (plaque formation units per ml.
Viral Infection:
Meanwhile one has to have freshly grown E.coli strains such as Y1089 (lysogenic) or Y1090 (lytic). These have to be grown in LB medium containing MgSO4 (20mM) and maltose 2%. The density of the cells to be at 0.5 to 0.7 OD.
LB medium:
10 gm Bacto tryptone,
5 gm Bacto yeast extract,
2% maltose,
20mM MgSO4,
Adjust the pH to 7.5.
Phage buffer:
200 mM Tris-Cl 7.4,
100 mM NaCl,
10 mM MgSO4
Add 100 ul of packaged phages to every 200 ul of cells and allow the phages to adsorb onto the cell wall surface. Keep the cells at 37^oC, then keep the cells on ice. Growing cells in Maltose produce substantial number of maltose transporters, which locate in the cell walls of the bacteria. Maltose transporter acts as receptors for the binding of lambda phage tail fiber.
Meanwhile one should be ready with 100ml of liquid made up of 1 gm Bacto tryptone, 0.5 gm NaCl containing 0.6% Agarose. About 2.5 to 4ml of this liquid is dispensed into 5ml tubes and they are maintained at 45^oC molten liquid.
Then the phage-bacterial mix is added to 2.5ml of liquid Agarose. At this point IPTG and X-Gal are also added. Swirl the tubes then over lay the liquid Agarose onto nutrient agar plates already prepared (LB media with MgSO4 and Maltose).
Incubate the plate at 42^oC for 4 to 5hrs then grow them overnight.
Plaques can be seen. The white plaques are those, which contain inserts, and those of blue contained self ligated DNA.
The plates can be used for screening the library with antibodies or DNA as probes as described earlier.
Plaques are seen with blue color or colorless. Colorless plaques are the ones that contain inserts and the colored ones are self-ligated vectors. Efficiency of cDNA library depends upon the number of white plaques you have got. Greater the number clones greater is the chance of getting the desired gene.
Screening with antibodies:
Though the ligation into lambda DNA under the lac-Z promoter, it is hard to believe that all the eukaryotic cDNA inserted into the site are capable expression. If the vector used is gt10 the inserts will be in the middle cI gene at E.coR1 site. The inserts that are ligated can be in any one of the directions. If the vector used is gt11 the inserts at the E.coR1 site at the carboxyl terminal of the Lac-Z gene and if any protein is expressed it will be in the form of fused protein. On the contrary if the vector is lambda 11 containing Sfi I, E.co RI and Not I sites at the carboxyl end of Lac-Z gene, the cDNA can be cloned in a specific direction.
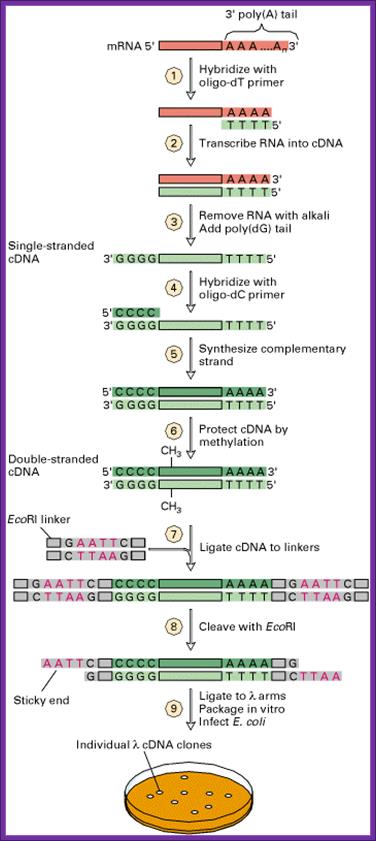
The inserts within the lac-Z gene can be screened as blue and white plaques. Similarly if GEM 2 and 4 vectors are used, cDNA can be cloned into one of the multiple cloning sites in directional manner and they will be under the control of a promoter. They cannot use for blue and white plaque screening and all of them appear as colorless plaques.
Whatever may be cloning site of the clones, if the cDNA are from eukaryotic systems expression is difficult to expect. If there are any expressions you are lucky. The reason for this is that bacterial translation system requires shine-Delgarno system for the initiation of translation while the EK mRNA has Kozak sequence for proper placement of mRNA at the ‘P’ site on the ribosome. Yet people have fancy notions, that there is a possibility of expression. The best bet to identify a clone in the cDNA library is to use a specific DNA or RNA probe.
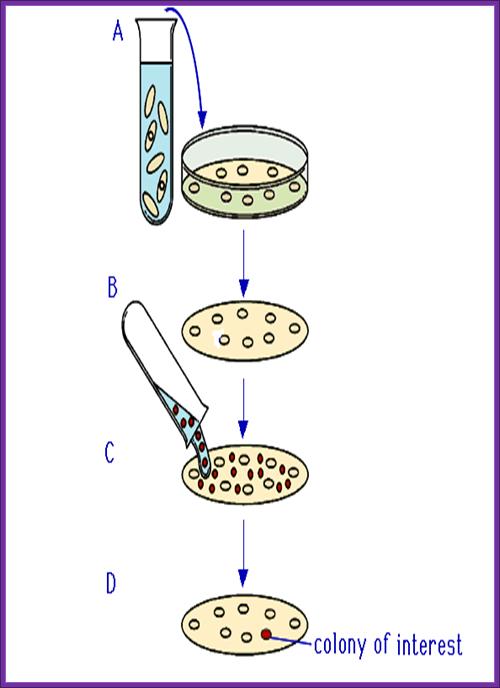
- Mark the positions in the plate and the plate numbers.
- Prepare NC membranes soaked in the buffer and place the same carefully without disturbing the plaques. Mark the positions on the membrane with reference to colonies in the plate.
- Keep the membranes for 10-15 minutes.
- Take out the membranes without disturbing the plaques and lay them upside down, so the colonies stuck to the membranes are on the upper surface of the membranes.
- Grow them over night or grow them to the pinhead size.
- Lyse the cells using nonionic detergents like noidet-50 or nonionic detergent like Tween with TBS buffer.
TBST buffer:
10mM Tris-pH 8.0,
150mM NaCl,
0.05% Tween or Nonidet.
- Surface dry but never bone-dry the membranes.
- Washing or dipping the membranes with the said buffer not only lyse the cells but also removes whatever agar granules are sticking to the surface of the membrane.
- Decant the TBST and add TBST buffer containing 2-3% skimmed milk, which, it is believed to block cellular proteins, so it is called blocking agent. Incubate the membranes for about 30 minutes on shaking water bath at room temperature.
- Wash the same 3 times.
- Then add TBST containing primary antibody. Note the antibody has to be quantitated and if desires it is to be dilute 1:200 times or more.
- Primary antibodies should be purified on DEAE cellulose columns or protein–A columns (protein-A is derived from staphylococcus aureus). This protein has a property to bind to primary antibodies strongly. Purification of antibodies is done first by incubating the primary antibodies in TBS buffer with E.coli extract overnight in cold. Spin to remove all the precipitate. This can be further purified by adding tissue proteins (general) to the antibody preparation.
- Such antibodies can be further purified by protein-A column or oligo-cellulose column.
- Such antibodies are aliquoted in TBS buffer and stored at -70^oC.
- Diluted primary antibody in TBST buffer is added to membranes and allowed to react for 2-5 hrs.
- Wash the free antibodies in TBS buffer two or three times.
- Surface dry the membrane and don’t dry it completely.
- Then add TBST containing anti-antibody conjugated with Alkaline Phosphotase enzyme. Incubate for 2-3 hrs and again wash the membranes in TBS two to three times.
- Decant and now add alkaline Phosphatase buffer containing NBT (nitro Blue Tetrazolium) and BCIP (5’Bromo-4’Chloro-3indolyl phosphate) and cove the plates with aluminum foils or black paper for 15 minutes.
Alkaline Phosphatase Buffer:
100mM Tris-Cl pH 9.5,
100mM NaCl,
5mM MgCl2.
- Wherever purple blue color spots develop they are the colonies that have expressed a specific gene product that one is looking for.
- This has to be further confirmed by picking out the colony and growing the same and immuno precipitating the protein.
Propagation of Recombinant Phage particles:
Once the plaque has been identified by an antibody or DNA or RNA probe, the plaque content has to be reaffirmed by screening the same plaque by the same probe.
In order to maintain and propagate the recombinant DNA, pick the identified plaque with a toothpick and suspend the same in100 ul of phage buffer or SM buffer or phage eluting buffer. The plaque contains both bacterial cells as well as released phage particles.
SM buffer:
50 mM Tris Cl 7.5,
100 mM NaCl,
8 mM MgSO4,
0.01%Gelatin.
Allow the phage particles to diffuse into the solution. Keep it overnight. Meanwhile grow LE 392 cells in LB medium containing 2% Maltose and 8mM MgSO4. Transfer 100ul of phage diffusate into 100ml of bacterial cells and allow cells to grow at 37^oC in orbital shakers for 3-4 hours. The liquid becomes cloudy. As more and more cells are infected and lysed the liquid becomes clear. Add 500 ul of chloroform to it and shake. Spin the liquid and take out the supernatant and isolate DNA by phenol: chloroform method. Other wise one can collect phage particles by adding 0.5 volume of 5M NaCl and 0.33 volume of 30% PEG 800 ( PEG dissolved in 1.5M NaCl). Mix well leave the solution over night in walking cold room and spin the ppt to collect phage particles. Then isolate DNA using Phenol-chloroform method.
The other method is picking the entire plaque with a toothpick and suspends the cells and phages in 1ml of phage buffer, keep it overnight. Grow Y1090 cells overnight. Add 60 to 100 ul of the diffusate to 0.1 ml of freshly grown Y1090 cells and allow page particles to adsorb to bacterial cells. Keep the culture for 20-30 minutes at 37^oC. Then take 2.5ml of the phage infected bacterial culture and overlay on nutrient agar containing Maltose and MgSO4 and allow them to grow and propagate overnight at 38^oC. The next morning one can observe lot of plaques. Then overlay 2-3 ml of SM buffer. Incubate the Petri plates at room temperature in a horizontal shaker for 2-3 hrs. Decant the liquid and add chloroform to 0.3volumes. The supernatant can be used for isolating DNA.