Chromosomal Nature-Before, During and After Gene Activation:
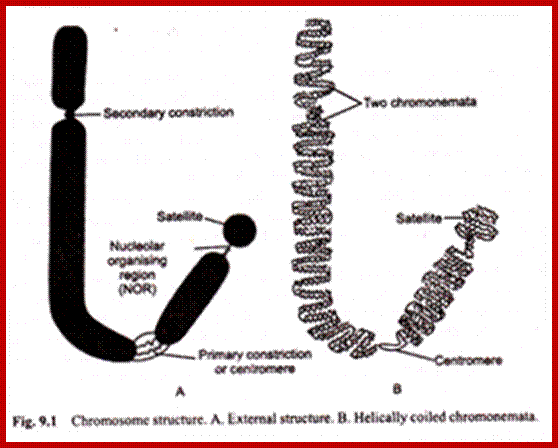
Basic chromosomal structure:
Chromosomal DNA of eukaryotes is linear, double stranded and very long ranging from 255Mb to 28Mb per chromosome. Human haploid DNA is 3-3.2x10^9 bp long, but compacted into 22 + X and Y chromosomes. Any such long DNA free from any structural support gets broken during replication, recombination and transcription. That is the raison d’etre eukaryotic systems have designed to compact such long DNA in to compact threads called chromosomes. Chromosomal DNA is associated with histone and nonhistones proteins, where histones participate as structural components and provide strength and stability and protect DNA from shearing and breakage; nonhistones act as functional or regulatory components either in activation or repression of genes.
Chromosomes-human; http://y12hb.wordpress.com/
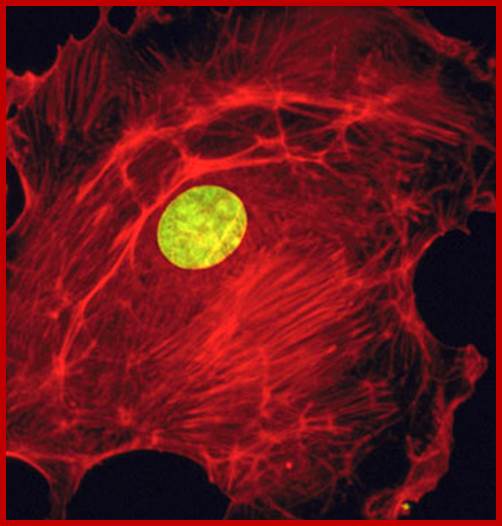
The Nucleus; fig.cox.miami.edu/
.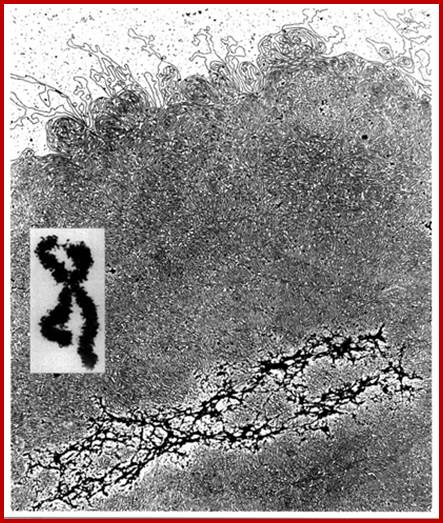
"Histone-depleted chromosomes (were studied) in the electron microscope. Our results show that: the histone-depleted chromosomes consist of a scaffold or core, which has the shape characteristic of a metaphase chromosome, surrounded by a halo of DNA; the halo consists of many loops of DNA, each anchored in the scaffold at its base; most of the DNA exists in loops at least 10-30 µm long (30-90 kilobases).'' Paulson, J.R. and Laemmli, U.K. Cell 12 (1977) 817-828


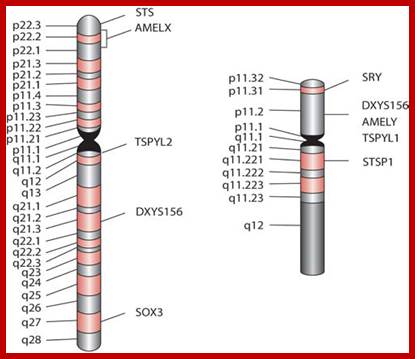
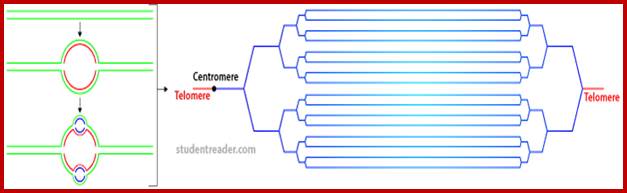
Composed of DNA and protein (histones) all tightly wrapped up in one package ;duplicated chromosomes are connected by a centromere; Amelogenin as a marker for sex identification in forensics and describe four additional Y chromosome markers, sex-determining region Y (SRY), Y-encoded testis-specific protein (TSPY), locus DXYS156, and steroid sulfatase (STS). The SRY, TSPY, DXYS156, and STS markers each have properties that could be used for developing more rigorous methods of testing forensic DNA samples for a Y chromosome or the presence of specific reproductive or secondary sex characteristics. http://www.avensonline.org/ http://www.uic.edu/; Applied Genetics-Chromosomes; http://on-line.ucol.ac.nz/genetics; http://www.apsubiology.org
http://www.biologydiscussion.com

Salivary gland PolyteneChromosomes; Balbiani);http://www.biologyexams4u.com/;http://www.biyanigirlscollege.com/; www.slideplayer.es/slide

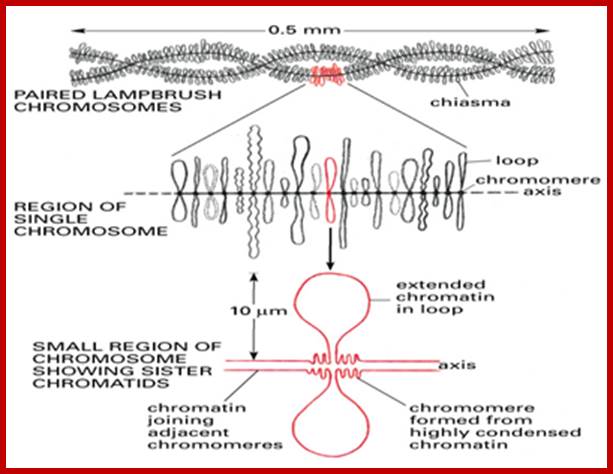
Lampbrush chromatid, central nucleoprotein produces chromatin loops which are transcriptional active; https://clinicalcenter.nih.gov
www.Keywordsking.com
Lampbrush chromosomes.

Chromosomes sequenced at the Sanger Institute. [Genome Research Limited]; http://webcache.googleusercontent.com/


Chromosome number, DNA content in BP and gene number per chromosome; SC = Secondary Constriction (NOR) in chromosomes 13, 14, 15, 21 and 22:
I
If you were getting excited about having a company like 23andme sequence your genome for you, it's time to put a lid on it. Apparently the State of California has decided that people should not be allowed to sequence their own genomes without supervision from a medical professional (despite the fact that many medical professionals are not trained to understand genomic data). It is weird remark. each of the chromosomes has two arms on either side of centromere. The smaller arm is called P-arm and the longer arm is called Q-arm. Based on linkage studies each of these arms are subdivided in P1, P2, p3 etc; similarly Q arms. Chromosomes showed in the diagrams are metaphasic most condensed chromatins (chromosomes);NCBI, Genetic Review; http://www.ncbi.nlm.nih.gov/
|
N0. |
Base pair x10^6 |
No. of genes |
|
|
1 |
263 |
2769 |
|
|
2 |
255 |
1776 |
|
|
3 |
214 |
1445 |
|
|
5 |
203 |
1023 |
|
|
6 |
194 |
1261 |
|
|
7 |
183 |
1401 |
|
|
8 |
171 |
1410 |
|
|
9 |
155 |
952 |
|
|
10 |
145 |
1086 |
|
|
11 |
144 |
1042 |
|
|
12 |
143 |
1626 |
|
|
13 |
114 |
1347 |
sc |
|
14 |
109 |
477 |
sc |
|
15 |
106 |
821 |
sc |
|
16 |
98 |
915 |
|
|
17 |
92 |
1139 |
|
|
18 |
85 |
1471 |
|
|
19 |
67 |
408 |
|
|
20 |
72 |
1715 |
|
|
21 |
50 |
357 |
sc |
|
22 |
56 |
657 |
sc |
|
x |
164 |
1090 |
|
|
Y |
144 |
59 |
|
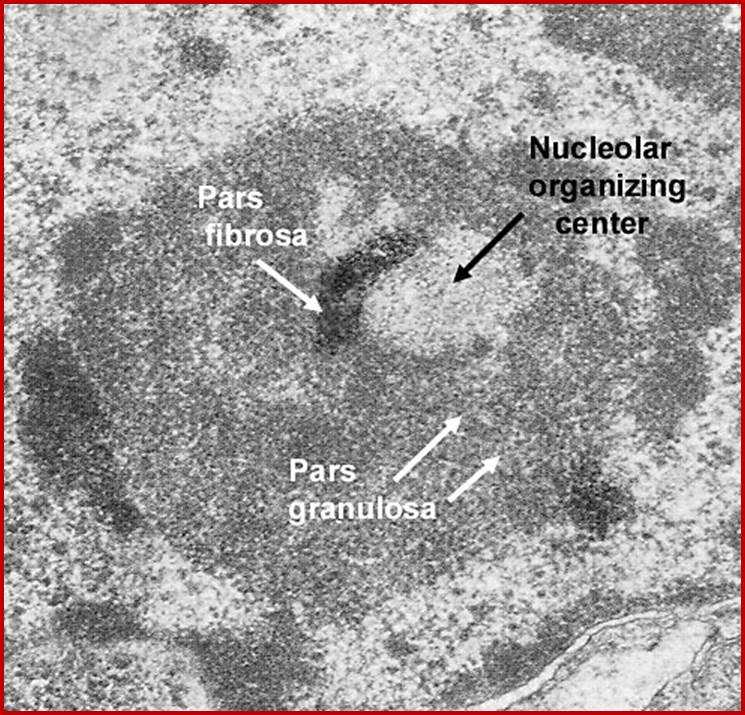
Nucleolar organizing centers (fibrillar centers): Pale staining regions containing DNA encoding rRNA,ii. Pars fibrosa: Electron dense fibrillar region composed of ribosomal RNA transcripts. iii. Pars granulosa: Granular-appearing regioncomposed of maturing ribosome particles.; http://dc304.4shared.com/
Nuclear structures- heterochromatin is bound to inner nuclear membrane matrix; the nucleolus shows different structures such as pars fibrosa and pars granulosa.

https://www.slideshare.net
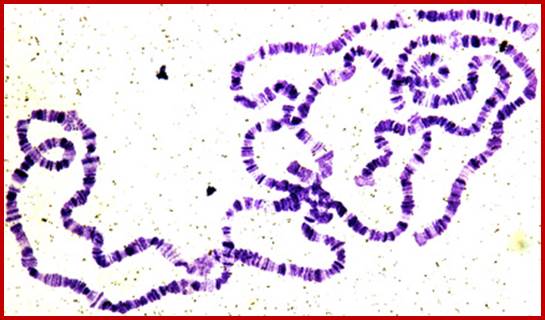
Metaphasic type of condensation is due to Structure (ral) Maintenance of Chromosome (SMC) proteins and non SMC proteins such as Condensins (contract the length) and Cohesins (glue two parallel chromatid strands). Differential staining with DAPI (4, 5-Diamino phenyl Indole) shows some darker bands and some lighter bands called G and R bands respectively; such bands can be discerned in compacted metaphasic chromosomes. Such dark bands are called heterochromatin and the lighter regions are called euchromatin; which exists in various states. Heterochromatin is classified into constitutive and facultative types; the first is found to be condensed always and the facultative locus varies from one tissue to the other. Constitutive heterochromatin is found at Pericentric and telomeric regions and Subtelomeric regions. The one of the two X-chromosome in mammals and many vertebrate’ cells is always heterochromatic. The metaphasic chromosomes are condensed 1400 fold in contrast to nucleosomal threads of 11nm thick. The metaphase dark bands contain more DNA per unit than euchromatin.
DAPI-4', 6-diamidino-2-phenylindole, dihydrochloride (DAPI) is a blue fluorescent DNA dye that targets double-stranded AT clusters in the DNA minor groove; The level of DAPI-DNA fluorescence is proportional to DNA content
Synteny maps (bottom map) for each Tetraodon (it is putter fish contains just 350MBps smallest among vertebrates) chromosome, colored segments represent conserved synteny with a particular human chromosome. Synteny is defined as groups of two or more Tetraodon genes that possess an orthologues on the same human chromosome, irrespective of orientation or order. Tetraodon chromosomes are not in descending order by size because of unequal sequence coverage. The entire map includes 5,518 orthologues in 900 syntenic segments. You can also do this in the other direction, and take each Tetraodon chromosome, color code them, break them apart, and reassemble them into the order they would be in the human genome, as in the second diagram.
Chromosome Painting Distinguishes Each Homologous Pair by Color:
Recently developed method for visualizing each of the human chromosomes in distinct, bright colors, called chromosome painting, greatly simplifies the distinction between chromosomes of similar size and shape. This technique makes use of probes specific for sites scattered along the length of each chromosome. The probes re labeled with one of two dyes that fluoresce at different wavelengths. Probes specific to each chromosome are labeled with a predetermined fraction of each of the two dyes. After the probes are hybridized to chromosomes and the excess removed, the sample is placed under a fluorescent microscope in which a detector determines the fraction of each dye present at each fluorescing position in the microscopic field. This information is conveyed to a computer, and a special program assigns a false color image to each type of chromosome. A combination of chromosome painting and fluorescent in situ hybridization, called multicolor FISH, can detect chromosomal transactions. The 24 types of human chromosome can be distinguished by different staining procedures. Each chromosome has a unique banding pattern, a distinctive pattern of dark bands (stained regions) and light bands (unstained regions). Banding of condensed metaphase chromosomes reveals about 450 different bands. Based on the banding pattern and the location of the centromere, chromosomes can be readily identified.
|
Banding: |
Overview: |
|
G Banding: |
G Banding (aka Giemsa Staining) is the treatment of chromosomes with trypsin (to remove chromosomal proteins) and then staining with Giemsa. Each chromosome pair stains in a distinctive pattern of light and dark bands. The dark bands are called G bands, and roughly correlate to base-pair composition (GC or AT) and repetitive DNA sequences. |
|
Q Banding: |
Q Banding involves staining a chromosome with Quinacrine mustard, and then examination by fluorescent microscopy. There are bands which brightly fluoresce and bands which are dim. The dim bands are Q bands, and correspond almost exactly to G bands. Q Banding is useful for detecting heteromorphisms. |
|
R Banding: |
If the chromosomes are treated (using heat or special chemicals) before staining, their dark and light bands are reversed. When G and Q bands stain poorly, then R banding is used to increase contrast provide better readability. |
|
C Banding: |
C banding stains centromeres, telomere and constitutive heterochromatin. Heterochromatin is a chromatin which remains condensed and stains darkly even in interphase (non-dividing) cells. |
|
Painting: |
Not actually a form of banding, chromosome painting involves hybridization of each chromosome using a chromosome-specific with a unique combination of fluorescent dies. The result is a colorful array of chromosomes, with each one painted a different color. |
High-resolution banding: It is G or R-banding of less condensed chromosomes, revealing 550 to 850 bands (as opposed to 450, as mentioned above). Usually involving prophase or prometaphase chromosomes, high-resolution banding helps pinpoint structural abnormalities.
Human Genome -3.9x10^9; distance between two bases=3.4^A; length of Haploid genome-~1 meter.
To pack DNA into the tiny nucleus (the DNA packing problem), DNA is tightly wound around special proteins to form a nucleoprotein complex called chromatin. Chromatin proteins are predominantly histones, a family (H1, H2A, H2B, H3 and H4) of small proteins that are conserved in eukaryotes and contain many positively-charged basic amino acids that interact with negatively-charged DNA phosphate groups.
Actively transcribed chromatin is in 10 nm form (beads-on-a-string). This can revert to 30 nm when genes are repressed. In interphase state one can observe differentially condensed and relaxed region of the chromatin which suggests active and inactive state of chromatin. Highly inactive chromatin, such as that containing highly repetitive DNA, is still further condensed around the chromosome scaffold. Histones have unstructured tails (not seen in crystal structure) that are specifically modified (including acetylation, methylation, and phosphorylation) to mediate the regulated condensation and decondensation of the chromatin.
|
Chromatin |
Overview: |
|
Heterochromatin |
Heterochromatin is made up of dense, tightly packed, portions of the chromosome that are mostly inactive and often contain repetitive simple sequence DNA. This appears very dark in the electron microscope. Heterochromatin (HC) is highly condensed but facultative (HC) can be converted to euchromatin by transcriptional activators targeted to that chromosomal region. Similarly euchromatin can be converted to FHC. |
|
Euchromatin |
Gene rich regions of the chromosome are much less densely packed and make up what is called Euchromatin. The decondensation of chromatin upon transcriptional activation can also be observed through its sensitivity to DNase. For example, globin genes expressed in erythrocytes (in these Cells, DNase sensitive) but not expressed in other cells. |
Basic chromosomal Chemistry:
The DNA is associated with histone and nonhistones proteins, where histones participate as structural components and nonhistones act as functional components either in activation or repression of genes.
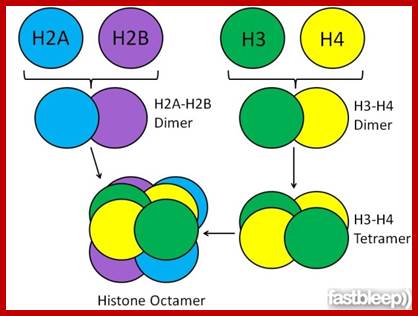
Basically there are tetramers of H3, H4 subunits and tetramers of H2A and H2B subunits; tetramerization is due to protein–protein interaction. The tetramers join with one another to form Octamers. All histones have ~3 helical domains as their histone folds, central long helix flanked on either side by smaller helices with loops in between.

Principles of Cell Biology; http://www.mun.ca/biology/
Composition of Histones:
|
Histone |
basic/acidic Ratio |
Mol.wt (kDa) |
Number of a.a |
Tail length-%R/%K |
|
H1 |
5.4 |
23 |
224 varies |
1 and 29 |
|
H2A |
1.4 |
13.96 |
129 |
23 ?9/11 |
|
H2B |
1.7 |
13.7 |
125 |
24? 6/16 |
|
H3 |
1.8 |
15.27 |
135 |
37? 13/10 |
|
H4 |
2.5 |
11.23 |
102 |
28? 14/11 |
Composition of histone amino acids; H3 and H4 are most stable ones than H2A and H2B. The most variable one is H1; There are some special histones variants of H3 such as CenA, CenB and CenC; Few special histones are found in specific cell types
Histones:
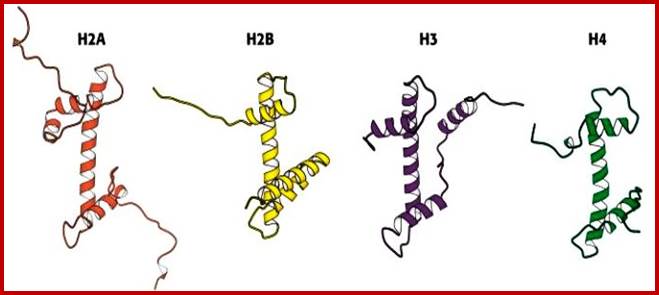
Simple diagrammatic representation of Histones H2A, H2B, H3 and H4 with their N-terminal tails; only H2A has tails at both ends of the protein; http://oregonstate.edu/
https://www.fastbleep.com

https://www.fastbleep.com

Histone Octamer; H2B,H3,H4and H2A + H2B,H3,H4 and H2A. http://www.carolguze.com/text

http://www1.biologie.uni-hamburg.de

The C-terminal blocks of Histones are called Histone fold, the sequence of a.a found in the N-terminal for they are involved modifications, they are called Histone Code. http://atlasgeneticsoncology.org

The N terminal sequences for Histones where amino acids get modified-called Histone code. http://eric.diluccio.fr/
All these histones are conserved, but Histone 1 is not that conserved, it varies from one specie to the other. H5 is a H1 variant. Histones in centromere are different and they called CENP histones.
Nucleosomal thread
Interestingly each of the histones has N-terminal tails with specific sequence of amino acids, but the H2A has c-terminal tail also. The helical motifs are called Histone folds and sequence of histone tails that are modified provide codes for the binding of specific proteins is often referred to as Histone code for they dictate the tail modifications at specific amino acid residues and compactness of chromatin.
Composition of Histones:
The histone octamer, as two tetramer group placed one above the other, are wrapped around by DNA in left handed mode and histone with their large number of positive charges bind non- covalently to DNA nonspecifically at least at 14 points. The length of DNA that wraps around is approximately 145-165 bp. Such a structural form is called nucleosome or Nu-body. The Nu-bodies (Octamers) are organized in a series of successive bead like bodies (beads on string); thus the chromosome, at the first order of organization, is called nucleosomal thread 11nm thick. Histone enwrapped with DNA is not static, but dynamic and it unwinds and rewinds constantly with some time gap and even histone octamer can slide along the DNA. This dissociation for that few seconds is more than enough for the protein factor to find their sequences. When the DNA wraps around the histone core, octamers, it assumes deformed state. DNA perse is right handed coiled state, but when it wraps around the histone octamers in left handed mode the DNA gets deformed.

Structure and assembly of nucleosomes; (a) View down the DNA helix axis of the nucleosome core particle from Xenopus at 2.8 Å resolution shows organization of the histones and DNA. The DNA strands (brown and green), are on the outside. The histone octamer forms a helical ramp, around which is wrapped 1.7 turns of a DNA super helix. Each histone consists of a three-helix domain called the ‘histone fold’, together with two unstructured tails. Pale blue hooks indicate points of DNA–histone contact. (b) Model of chaperone-assisted nucleosome assembly during DNA replication in eukaryotic cell nuclei. CAF-1 binds a histone H3–H4 tetramer (blue and green) and docks with the PCNA clamp on replicating DNA. NAP-1 binds a H2A–H2B dimer (red and brown) and transfers it to the docked histone tetramer. The model is purely schematic and does not indicate precise interactions or topologies. Recent work has clarified the role of nuclear chaperones in the assembly of nucleosomes. Part (a) reproduced, with permission, from Ref. [39]; part (b) reproduced, with permission, from Ref.. R. John Ellis
 Assembly
of nucleosomeshttp://www.mikeblaber.org/
Assembly
of nucleosomeshttp://www.mikeblaber.org/

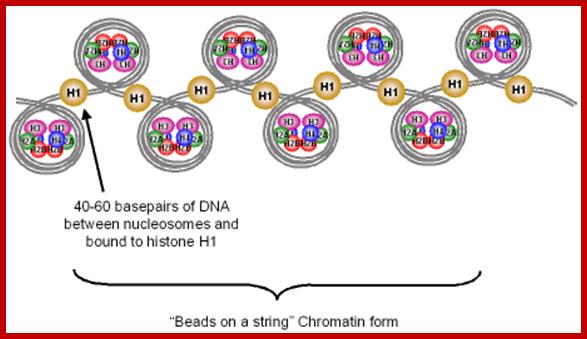
Nucleosomal thread 11nm; nucleosomes are held together by linker DNA; ;Abdul Rauf Shakoori; ;https://link.springer.com
The nu body is ~110A diameter and ~60A thick. The DNA wraps around the histone octamers by 1.65 turn in left handed super coil. The Nucleosomes are regularly placed with DNA in between two successive nucleosomes is about 50-90 bp called linker DNA. Histone-1 with its central globular and C and N tails binds to linker DNA found in the linker region in such a way it occupies a cavity like structure where DNA entry and exit around the histone is on the same side. The H1 tails also bind to linker DNA at either ends by 18-20 bases, so the length of the linker DNA shrinks and at the same time the cross binding of H1 induces torsion such that this leads to compaction which may assume solenoid coils or zigzag form of 30nm size; this is the basic chromonema form. Both 11nm and 30nm threads are used for gene expression. Even bacterial DNA gets compacted with binding of histone like proteins and generates loops with protein bound bases.
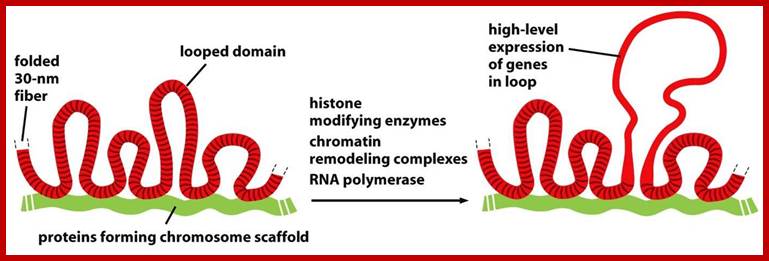
Experimental demonstration of chromatin loops in interphase chromosomes (NCBI/NIH); In situ hybridization of interphase cells was carried out with several different fluorescent-labeled probes specific for sequences separated by known distances in linear, cloned DNA. Lettered circles represent probes. Measurement of the distances between different hybridized probes, which could be distinguished by their color, showed that some sequences (e.g., A, B, and C), separated from each other by millions of base pairs, appear located near each other within nuclei. For some sets of sequences, the measured distances in nuclei between one probe (e.g., C) and sequences successively farther away initially appear to increase (e.g., D, E, and F) and then appear to decrease (e.g., G and H). The measured distances between probes are consistent with loops ranging in size from one to four million base pairs. http://www.pha.jhu.edu/ [Adapted from H. Yokota et al., 1995, J. Cell Biol. 130:1239.]; slideplayer.com/slide/; https://www.studyblue.com
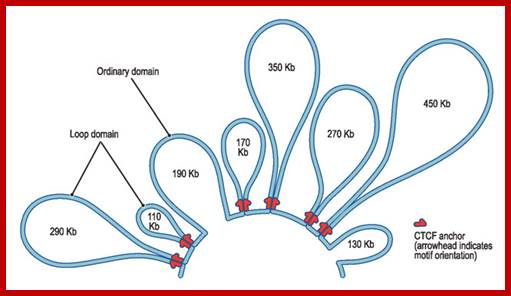
Genes sit on a loop right near the large regulator molecules needed to start and stop their production (promoters, enhancers and repressors). Loops can be flexible and the contact of the sites can be intermittent. This loop region makes it much easier to use the DNA. Often these loops create the environment for the activity, but a further stimulus is, also, needed. Chromatin loops; http://jonlieffmd.com/
Each type of chromatin structure works to form regions with TADs. Some factors keep the different types apart, making them more localized. TAD-topologically Associated Domains; http://jonlieffmd.com

Human mitotic chromosomes stained to reveal a scaffold-like structure along the chromosome axis
In these confocal fluorescence micrographs, the DNA has been stained with a blue dye, and the axis has been stained red with a fluorescent antibody against a protein in the condensin complex. Only part of the scaffolding is visible in these optical sections. (A) A typical mitotic chromosome, which has a gently coiled scaffold along each of the two chromatids. (B) A metaphase chromosome from a cell artificially blocked in metaphase; in the chromosomes of these cells, the scaffold has condensed by further helical folding. A 2M NaCl extracted scaffold protein is fibrous with molecular weight of 37kDa and 83(85)kDa. The scaffold protein is also associated with Topoisomerase II (140kDa). Perhaps the most abundant chromosomal nonhistoe protein may be TopoisomeraseII. It can expand and contract. (Courtesy of Ulrich Laemmli and Kazuhiro Maeshima).
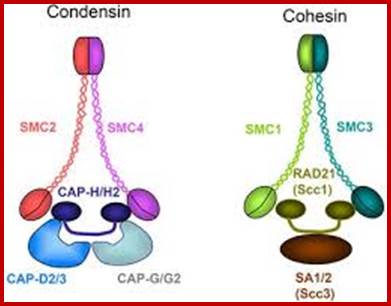
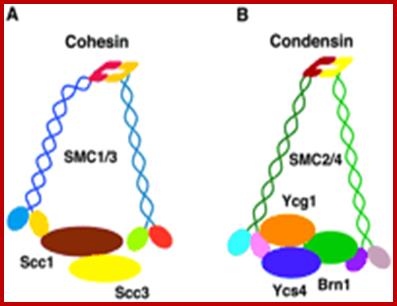
Sister chromatid cohesion is mediated by entrapment of sister DNAs by a tripartite ring composed of cohesin’s Smc1, Smc3, and α-kleisin subunits.Condensins consists of SMC2/CAP-E and SMC4/cap-c, plus they also contain few associated proteins, cohesins glue chromosomal threads and condensins contract the chromosomal thread;http://mcb.asm.org/http://openi.nlm.nih.gov/

Condensin I associates with structural and gene regulatory regions in vertebrate chromosomes; The condensin complex is essential for correct packaging and segregation of chromosomes during mitosis and meiosis in all eukaryotes. We find that condensin I binds predominantly to promoter sequences in mitotic cells. Ji Hun Kim, etal; http://www.nature.com/

http://www.valdosta.edu/~jfelder/molecular1.ppt ;http://edu.docdat.com/

The central scaffold protein is visible in lampbrush chromosomes of oocyte cells and one can observe the opened loops of various lengths, which are all active in transcription; each loop can be considered as one chromomere; from the extension one can measure the length of DNA of each chromomere. http://earthkart2011.blogspot.in/
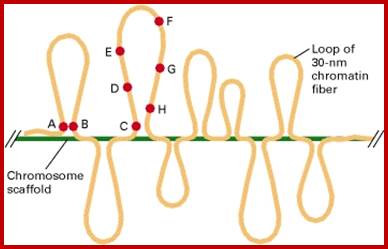
Nucleosomal thread of 11nm thickness; it is the basic chromosomal form; this form exists in certain regions of the chromatin at interphase. The third level organization of chromatin is that the 30 nm fiber associated with various nonhistones proteins, among them Topoisomerases and HMG proteins are found in large numbers. Histone depleted chromatin shows a long structural protein runs the entire length of chromatin called scaffold protein and the chromosomal DNA is found looped out of such structure; the base of the loops is attached to the scaffold; the loops are of various sizes 20 to 90kb (15 to 30um). A cross sectional view of a typical chromosome consists of a central scaffold protein from which histone bond DNA threads (coiled) appears to loop out; thus the thickness of the structure is 1um, it agrees with the thickness of the intact but relaxed chromosome.
Chromosomes with 140 -250million base pairs could easily produce about 2000 -3000 such ~70kb long loops. The base of DNA loops containing AT rich sequences are bound to scaffold protein complex; such regions of chromonema are called Matrix Attachment sites (MARs or SARs scaffold attachment). MARs/SARs are AT-rich DNA sequences, often containing topoisomerase II at the base that mediate the anchoring of the chromatin fiber to the chromosome scaffold or nuclear matrix and might delimit the boundaries of discrete and topologically independent higher order domains. The scaffold proteins can be associated with ScII 85KDa; they have ATPase domains. Chromatin SMC proteins also play important role chromatin organization.
Interestingly interphase chromatin is also associated with several TFs bound to specific regions of the DNA coils. Interestingly the base can also associate with histone deacetylases or Acetylases. DNA associated activators (or TFs) or repressors at specific positions throughout the cell cycle is a fact; if one finds such proteins associated with chromatin, provide the specificity of the gene loci. The chromatin compaction leads to 300nm thickness. This further compacts to 700nm to 1400nm thick at metaphase. The 700nm compaction is due to coiling of 300nm loops. Such spiral coiling facilitates the opening of chromatin easily. At the same time these threads are strengthened by scaffold and non-histone proteins.

Nucleosomes folded and compacted into a bundle of fibers; http://www.studyblue.com/

New structural model for the metaphase chromosome based on thin plates; Experiments performed using several different microscopy techniques have allowed researchers at the UAB Chromatin Laboratory to discover that, during cell division, chromosome DNA is packaged within planar structures formed by many extremely thin layers. These planar structures are stacked, occupy the entire volume of the chromosomes, and are probably oriented perpendicular to the central chromatid axis. The planar geometry of these structures is very well defined, but the nucleosomes inside the successive layers are irregularly oriented. Pablo Castro-Hartmann, et al;http://www.uab.es/
“We (above authors) have discovered that in condensed chromosomes, chromatin is densely packaged forming plate-like structures instead of the typical fibers considered in the current models of metaphase chromosomes. Our electron microscopy images have shown that chromosome plates can form multilayered structures, having a thickness of approximately 6 nm each layer”.

Chromo DNA to metaphasic Chromatin; http://en.wikipedia.org/wiki/Chromatin.
When chromatin duplicates at S-phase, the replication fork drives through ds DNA separating parental strands into leading strand and the other as lagging strand. As the strands separate, the histone octamer randomly associate with single strands distributed equally among them. During replication, as new dsDNA produced gets associated with histone octamers. Histones synthesized at this point are more or less acetylated and the same organize into H3x2 and H4x2 tetramers and bind to dsDNA and then H2Ax2 and H2Bx2 join. Most of the histone tails are acetylated at specific amino acids. The assembly is facilitated not only by acetylation of H3 histones but also by Nucleoplasmin; and probably Topoisomerases assist in the formation of new chromatin threads; nucleoplasmins are considered as Chaperonins.

Model of the chaperone function of Nucleoplasmin: (a) Face views. (b) Side views. The nucleoplasmin pentamers (blue and yellow) dimerize to form decamers, a process that might be favored by the binding of H2A–H2B dimers (red). The formation of decamers, either alone or in the presence of H2A–H2B dimers, might trigger a conformational change in the pentamers such that the β hairpins (blue and yellow) become extended outwards. The dimers bind to the β hairpins and A1 tracts of the nucleoplasmin decamer. An H3–H4 tetramer (green) then binds to each dimer to form an octamer. This binding is repeated five times on the lateral surface on the decamer to form a decamer–octamer complex. www.cell.com


https://www.slideshare.net
“Each extended loops as shown in the upper diagram looks like a knob like projection at the surface and it is believed to represent the tip of a separate looped domain. Note that the two identical paired chromatids in the diagram can be clearly distinguished. (From M.P. Marsden and U.K. Laemmli, Cell 17:849-858, 1979. © Cell Press.); “organization of these loops looks like Zea mays grains on its central axis in the cob” The binding of DNA to central scaffold provides strength to 30nm fibers it does not interfere with replication of DNA, recombination or Repair”. The composition of scaffold protein is still to be understood.
Yet one can observe metaphase chromosome perse certain regions where the chromatin is more compact, in Pericentric and telocentric regions. These compactions are due heterochromatization which can be distinguished by staining method. Telomeric HC is due to RAP1 and Sir Proteins, where Sir2 is histone deacetylases. Deacetylation can lead to binding of more deacetylases activity and more binding of proteins such as RAP1 and SiR. But the pericentromeric heterochromatization is due to small micro RNAs such as siRNAs as well as histone deacetylation, methylation of histone H3K9 and K27; and HP1binding to them. Heterochromatization spreading in euchromatin and other region is blocked by specific DNA sequence based protein subunits called insulators.
Chromatin Insulators and Enhancer-blockers
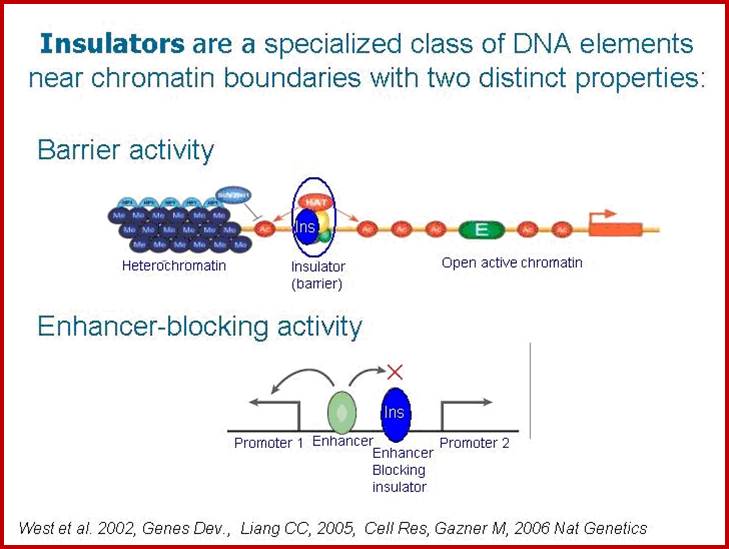
“The ability of chromatin to organize into functional autonomous units characterized by specific levels and patterns of expression is ensured through the establishment of boundaries that delimit these domains. In some cases, transcriptionally active genes may be embedded in an environment containing extensive regions of condensed chromatin capable of inappropriately silencing their expression. In other cases, signals from extraneous enhancers could cause incorrect pattern of expression of silent genes located nearby. Boundaries are responsible for ensuring the maintenance of the appropriate level of expression of each gene or gene cluster by marking the barrier between chromatin domains of distinct states.
These boundaries might occur at various positions, as a result of a balance between countervailing processes (such as chromatin condensation and decondensation), or be fixed at specific DNA loci. Chromatin insulators are DNA elements that mark the boundaries of chromatin domains by limiting the range of action of enhancers and silencers and by preventing incursions of neighboring chromatin domains. Although they have a wide variation in DNA sequences and proteins that bind to them, they are characterized by at least one of the following abilities; enhancer-blocking or barrier activity”.
A chromatin insulator with enhancer-blocking properties is able to block the enhancer-promoter communication when positioned between them. Enhancer-blockers restrict the long-range activation potential of eukaryotic enhancers in order to strictly limit their influence to one or few specific target promoter.
“The processivity model, envisions the relay of the enhancer signal to the promoter as a tracking action along the chromatin fiber. That model corresponds to a protein-tracking model in which the RNA-polymerase II-complex assembled at the enhancer tracks from the enhancer along the DNA to reach the promoter and activate mRNA synthesis. The transmission of this type of signal could be disrupted by the interposition of the insulator nucleoprotein complex interposed between the enhancer and the promoter”.
“In the topological model, instead of sending a signal from afar, a distant enhancer has to be brought close to the promoter through mechanisms that allow direct interaction between the enhancer and the promoter (e.g. by loop formation). Insulators may recruit chromatin-modifying enzymes to locally change the chromatin state disabling loop formation and thus preventing the direct enhancer-promoter interaction”.
“Barriers Elements: Barrier elements are none other than insulators and they have been shown to be able to recruit histone-modifying enzymes, locally competing with the spreading of silent chromatin markers often found between active genes prevent the spreading of gene activation to the neighboring gene improperly”.
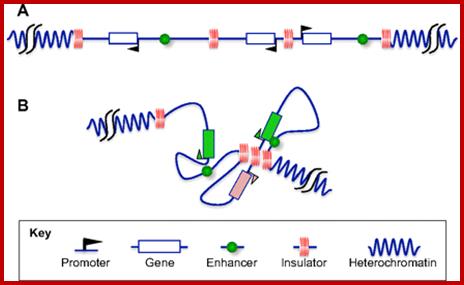
Specifically, an insulator may have an enhancer-blocking activity or a barrier activity. Here is an illustration of these two functions of insulators: ;http://www.anti-agingfirewalls.com/
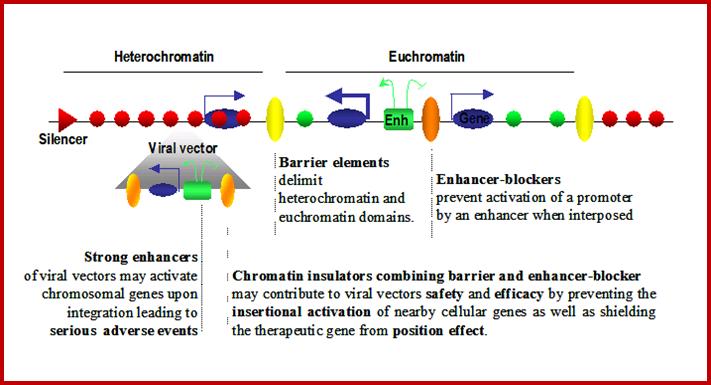
Eukaryotic Genome Organization
Legend: Eukaryotic genomes are composed of chromatin harboring in different states: Heterochromatin corresponds to closed chromatin, non permissive for gene expression, highly dense in nucleosomes bearing markers of silent chromatin (red circles). In this domain, silencers (red triangles) are responsible for limiting genes’ transcription level (thin blue arrows). On the other hand, euchromatin domains are composed of nucleosomes bearing marks of permissive chromatin (green circles) and contain highly expressed genes (thick blue arrows). Enhancers (green squares) are driving the expression of these genes and are capable of activating genes over large distances (green arrows). Chromatin insulators secure the delimitation of chromatin domains by limiting the spread of silent chromatin (barrier element, yellow circles) and also restrict the long-range influence of enhancers (truncated green arrow) when interposed between them and promoters (enhancer-blocker element, orange circle). Viral vectors (grey triangle) flanked by effective chromatin insulators, combining both enhancer blocking and barrier properties, raise the prospect of safer and more efficient gene therapy vectors.
Alan Cohen etal; http://www.nyas.org/
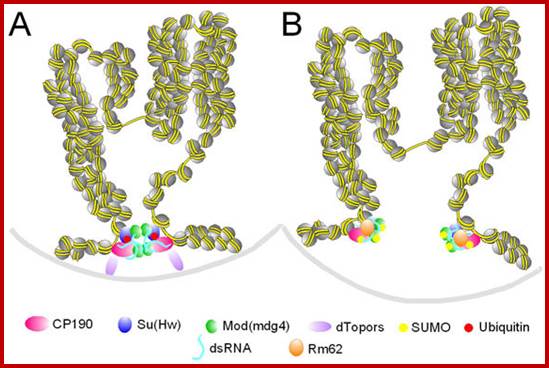
http://emboj.embopress.org
Insulator activity can be regulated by ubiquitination and sumoylation of insulator proteins. A. Two active insulators coming together at an insulator body. dTopors is present at the insulator sites, Mod(mdg4)2.2 and CP190 are not sumoylated and dTopors serves as a bridge to the lamina. B. Two inactive insulators that cannot be part of an insulator body. dTopors is absent and Su(Hw) is not ubiquitinated, whereas Mod(mdg4)2.2 and CP190 are sumoylated. The two insulator sites cannot interact with the lamina or each other and form insulator bodies

Chromatin domains and insulators; http://wwwuser.cnb.csic.es/
Insulators also block the effect of repressor spreading. A transgene (represented by gold DNA) integrated in the chromosome in a region of condensed chromatin is not properly expressed; the repressive chromatin structure of the surrounding region presumably spreads into transgene sequences, inhibiting enhancer-promoter interactions. B. If the transgene is flanked by barrier insulators (red DNA with two proteins represented as dark blue and green spheres), these sequences inhibit the spreading of the repressive chromatin, allowing an open chromatin and normal transcription of the gene.
Nucleating Centers in the Chromatin:
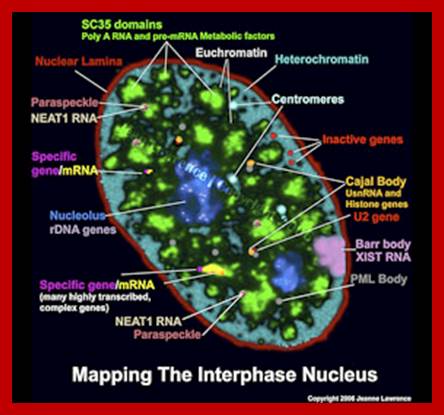
Interphase Nucleus full of active components
Gene expression starts with assembly of Basal Transcription Associate factors or apparatus called BTA factors in sequence, it need not be, but discerned by experiments; TFIID binds to TATAA site, how it is identified? It is identified by the sequence and nucleosomal acetylation region; this provides the site for nucleation. It is not a chance but it is determined by TBP to their specific DNA sequence TATAA this acts as nucleating center for assembly of other Transcriptional complex. The assembly of the BTA is complex but once in its place they can interact with upstream elements, whatever they are, whichever they may be, and by conformational changes in the protein complex induce the opening of dsDNA into transcriptional bubble, which is required for ensuing transcription. In another way transcriptional initiation can be possible by the binding of upstream factors in sequence specific manner and recruiting other components so as to modify histone tails by acetylation and loosening the nucleosomal structure to facilitate the binding of the rest of Transcriptional complex downstream. Loosening of the chromatin structure makes the promoter region to be freed from nucleosomes for the assembly of transcriptional factors, finally the RNA pol complex.
It is important to realize the promoter and regulatory elements in DNA should be made available free from proteins in the chromosomes. Before initiation of transcription; chromatin has to be remodeled in such a way at least some prime gene activating factors identify the said gene site (s) in sequence specific manner in the chromatin and bind and induce changes in chromatin structure. The same thing holds good for gene silencing or repression. There are remodeling protein complexes which are multisubunit structures; they are responsible for remodeling the chromatin so as to make the specific DNA sequence made available for the transcriptional apparatus assemble or not made available for the binding of transcriptional complexes. Perpetuation of chromatin status, active or inactive is an intrinsic process and it is executed precisely and exactly, until other factors are innovated to change the status.
Chromosomal State During Gene Expression:
Regulation of gene expression in eukaryotes is more complex and intrinsic. The genome is organized into a nucleoprotein complex of different orders. Chromosomes bear genes of different types such as protein coding mRNAs; NC RNAs such as rRNA, tRNA, RNAi’s (Si RNAs and MiRNAs), ScRNAs, SnRNAs, SnoRNAs, activator RNAs, TmRNA, antisense RNAs, Riboswitch RNAs, Lnc RNA, Linc RNAs, X-inactivating RNAs, TasiRNA, RasiRNA, PiRNA, telomerase RNA, RNaseP RNA, 7sK RNA, 7sLRNA and many more which are expressed differentially during development and also in tissue specific manner during and after development. Even after development genes are expressed in tissues in response a variety of signals.
Preferential association between co-regulated genes at Transcriptional factories;
Chromosomes undergo structural changes, has been observed, during cell cycle, from relaxed state at interphase to highly condensed state at metaphase. At interphase substantial numbers of genes are expressed in tissue specific manner. At metaphase the chromosomes are condensed to such an extent, all genes in them are shut off. Completion of cell division leads to chromatin to relax and transcribe the required transcripts for the cell.
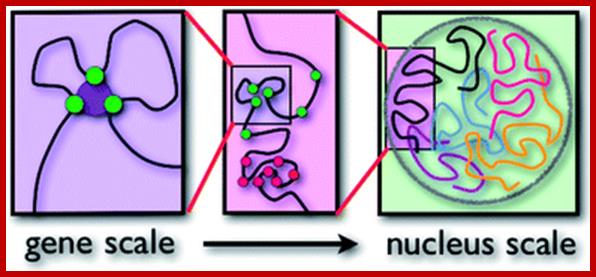
It is during this process chromatin relaxes and positions in nuclear milieu and get attached to the inner surface of the nuclear membrane associated nuclear matrix proteins through their heterochromatin loci. Even in response to signals, for cell division, cells acquire inputs for cell determination and differentiation. In the interphase whatever the cell types; the relaxed chromatin, certain regions or loci loop out into nucleoplasm. This region of the chromatin of 11-30nm thick, now it is accessible to transcription complex; it does not mean that all those genes present in euchromatic region are expressed, it is not so. The looped euchromatin DNA that is engaged in transcription exhibit what is called ‘transcriptional factories’ where active loops from different domains of chromatins of different chromosomes are clustered together where one finds active transcription of different set of genes located.
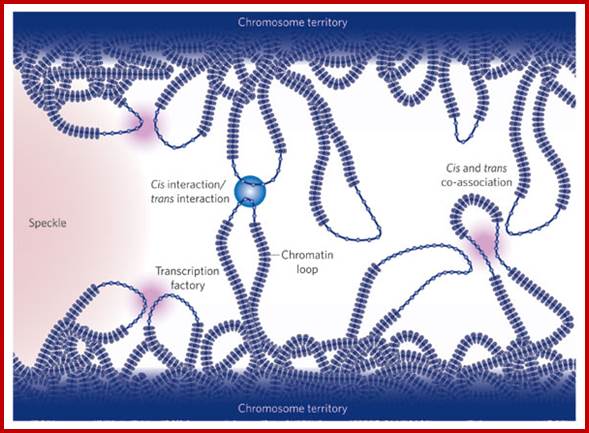
Colocalization of genes in the nucleus for expression or coregulation.Peter Fraser & Wendy Bickmore;http://www.nature.com/
Active genes on decondensed chromatin loops that extend outside chromosome territories can colocalize both in cis and in trans at sites in the nucleus with local concentrations of Pol II (namely transcription factories; dark pink) and adjacent to splicing-factor-enriched speckles (pale pink). Interactions can also occur between regulatory elements and/or gene loci and lead to coregulation in trans (blue circle).
![]()

Long range chromatin interactions
Genome organization in mammalian nuclei: Chromosome conformation varies between cell types and this inevitably places whole groups of genes in particular nuclear environments, such as regions in the nuclear interior that are rich in splicing factors; or next to transcription factories, where many RNA polymerases simultaneously transcribe different transcription units. Genome architecture within the interphase nucleus is inextricably linked with gene regulation.

Active genes (red) with transcription factories (green) in T cells.

Lamin Associated Domains LADs may consist of relatively condensed chromatin (thick lines) and aggregate at the nuclear lamina. Other repressed regions may interact with each other in the nuclear interior, as do active regions. Complexes formed by components of the transcription machinery (transcription factories) and CTCF may tether active regions together. Parts of only two chromosomes are depicted, each in a different color for clarity. Most interactions occur within chromosomes, and relatively few occur between chromosomes; Bas van Steensel & Job Dekker; http://www.nature.com/
It is logical to expect that chromosomal loci where gene to be expressed requires unwinding and the nucleosomal structures are relaxed or at least some part of the DNA of the said gene is to be free from histones for the binding of nucleating pioneer proteins, which recruits histone acetylases and transcriptional complex and its related factors.
Whether the regions that are active in gene expression or not can be tested by DNase1 or micrococcal nuclease treatment, which on partial digestion of the chromatin DNA wherever it is free from proteins is digested and the same can be analyzed on gels as several bands of uniform sizes. Nucleosome bound regions show ladders and nucleosome free regions as blanks.
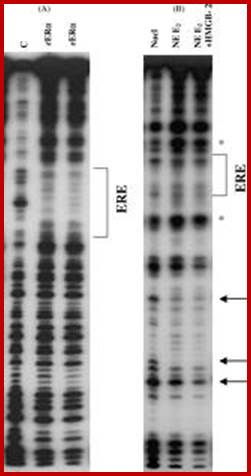
DNase-1footprint analysis of ER binding to probe DNA is different from binding to mono-nucleosomes. (A) DNase I footprint with labeled probe DNA (ERE) in the absence or presence of 2 and 4 μg recombinant human ERα. After DNase treatment reaction products were run on an 8% acrylamide-8M urea sequencing gel. The bracket indicates the position of the consensus ERE and protected sites. (B) DNase I footprint with labeled mono-nucleosomes in the absence or presence of 20 μg ER-containing nuclear extract plus or minus 10 μg HMGB-2. Arrows indicate sites of enhanced cleavage of DNA bases with addition of ER ± HMGB-2 vs. absence of ER. The symbol (*) indicates sites of decreased cleavage.
This can be observed by gel electrophoresis. So the DNA of a gene that is active state should be free from nucleosomes, and mostly it is of promoter regions; it is the promoter region where transcriptional apparatus and its associated factors bind upstream of InR and inclusive of InR. The most fascinating aspect is what makes the DNA of gene or genes accessible or not accessible for transcriptional initiation and transcription? The simple explanation is that when histones are free from promoter region of DNA, the promoter of the genes accessible but if it is bound it is not. What makes the histone to be freed form such regions? Is it due to the binding repressor to DNA that perpetuates that makes the promoter not accessible or DNA is tightly compacted by specific proteins so it is not accessible. For the DNA to be free such chromatin compacting proteins should be freed. The crux of gene activation and gene inactivation is in understanding of chromatin remodeling during the process..
State of chromatin when Gene Expression is on Grand scale:
1. Ribosomal RNA gene expression:
All eukaryotic cells irrespective of species, do exhibit rRNA synthesis all the time in the nucleolar region of the nucleus. Nucleolar size changes, when pericyclic cells are activated, in the case of IBA induced root initiation in Phaseolus vulgaris, the size of the nucleolus is so large it occupies ¾ of the nucleus. This shows the requirement of rRNA for pericyclic cell not for others. Requirement of rRNA for any cell is very high and so requires the transcription of rRNA all the time and in large amounts. To supply such high quantity, 100-200 rRNA genes in tandem repeats in the secondary constriction region, looped out as naked DNA, depending upon the species and the stage of development. In humans chromosome 13, 14, 15, 21 and 22 contain rRNA genes in a region called NOR or nucleolar organizer region. After telophase as the nuclear membrane reconstitutes, chromosomes relax and open; at this point of time rRNA coding DNA open out in the form of loops of various sizes and organize into nucleolus, where the DNA is freed from all chromatin proteins and gets associated with transcription complex and transcription goes on. And one can visualize each of the rRNA genes that are in the process of transcription, on the open DNA, and one can observe Christmas tree like transcripts arranged-from the nascent to old transcript. In this the chromosomal DNA is totally devoid of any histones, and the only proteins associated are RNAP I and its associated factors are found. This goes on 24 hrs a day and 365 days a year. This implies for massive transcription the DNA should be free for the transcription complex to operate.
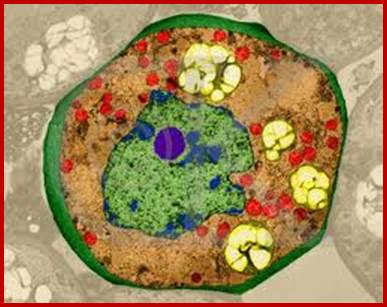
Nucleus is green in color and nucleolus in blue color
![]()
It has been observed nucleolar loci exist in certain chromosomes and not in all chromosomes. They are inherited by the same parent, i.e. but pollen-male and egg-female are contributor; it is virtually similar to biparental contribution. This is true for biparental off springs. It so happens, the nucleolar DNA that opens up for transcription is either from one parent or the other. So one set of rRNA genes are kept silent and the other is expressed; which is similar to X chromosome inactivation.
http://www.indiana.edu
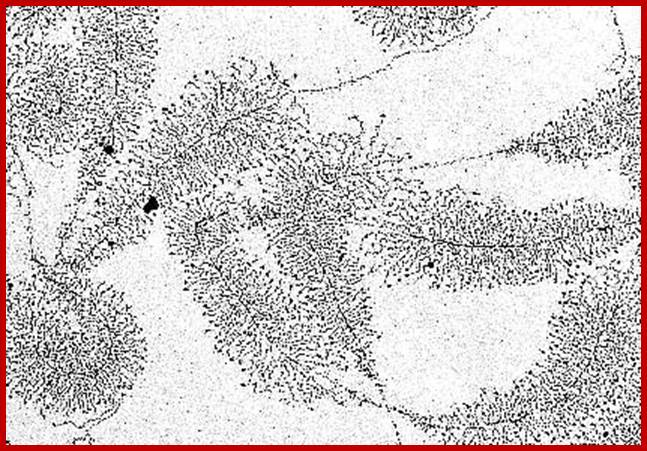
rRNA genes exist in tandem arrays. Each of the genes contains core sequences and upstream promoter-enhancer components. The rDNA is transcribed in very high quantities and higher rates. As a series of RNAP I assemble at the start point and initiate transcription and end; one can observe the transcripts from short to long ones, looks like Christmas tree pattern. Note not all rRNA gees are expressed; only 40 to 50% of them are expressed; In the nucleoli of the oocyte nucleus of Triturus virescens, an American newt species (as well as with other amphibians) occurs free DNA. The picture shows the transcription of genes that are carriers of the information about ribosomal RNA formation ("MILLER-trees"). More about the interpretation of the electron microscopic picture (O. L. MILLER, B. R. BEATTY, Biology Division, Oak Ridge National Laboratory, http://www1.biologie.uni-hamburg.de
2. Lamp brush chromosomes:
Another system that shows such grand scale transcription at a particular stage of development is Xenopus oocyte stage. During meiosis at pachytene-diplotene transitory stage, one finds meiotic chromosomes are maximally elongated and one can observe large number of granular structure all along the length of each synaptically paired chromonemal threads, at some points one can observe chiasmata also.

Top figure, Lampbrush cromosomes active in transcriptionhttp, diagramtic features of structural organization of Lampbrush chromatin http://www.nptel.ac.in/
Chromosomes contain chromomeres (cytological observation), which are nothing but coiled-coiled compacted chromonemata containing clusters of genes. Some chromomeres in such synapsed chromatin are opened out into naked DNA loops of 5kbp to 50kbp long. Such loops are free from chromatin associated histones. Nearly 5000 such loops have been counted. In between such opened or say active chromomeres there are chromomeres compacted into inactive chromatin. Such looped out DNA is free from histone complexes and found to be actively transcribing. Such kind of transcription is required for the future development of egg into an embryo after fertilization. This is a preparatory stage where the developmental process requires proteins, rRNA, mRNAs and many others in massive quantity. They are produced and stored.
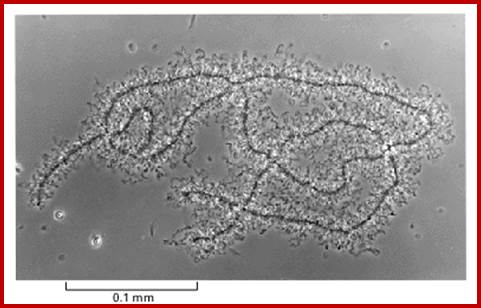
3. Salivary gland chromosomes:
Another grand scale expression is seen in insect larval development like drosophila and its related species. Such a scenario is also observed in plants’ haustoria during the development of plant embryo. Developing plant embryo requires all such inputs for the embryo development and it is provided by the polytene chromosomal formation and gene expression on grand scale or say large scale.
When the fertilized egg develops into larva, in the case of Drosophila, on reaching 11th day it enters into pupa stage. There is a dramatic transformation of the larval body into pupal structures and later into insect per se. This huge transformation requires a large scale expression all those genes involved in metamorphosis. The larval cells have four pairs of chromosomes and the total number of genes in the insect is about ~18000 or so. The homologous chromosomes are found paired, as if it is meiosis. The number of genes’ expression required for such transition has been analyzed by micro array chips and found to be in large numbers. But most of the genes exist as a pair of genes on their respective homologous chromosomes. But the requirement in such in that period is huge. In order to supply to such demands, each of the required genes has to be expressed at very high rates in the transitional period; this looks like an impossible task. But large scale expression made possible by chromosomal DNA duplication into ~1080 chromonemal strands irrespective of what genes this is are expressed what genes remain silent. Thus each gene is represented ~1080 times. The gene expression at each loci amounts to thousand genes that suffices the demands. So insects have designed that their chromosomes in Salivary glands on 11th day undergo transformation into multistranded, visible under normal microscope, this is differential gene expression at cytological level. Starting from the early 11th day larva till the pupal initiation, sets of genes are expressed temporally, it means at the early stage one set of gene are expressed, as the development progresses, another set of genes start expressing and the early genes expression regresses. By the end of this progression of thousands of individual genes are expressed and thousands become silent a cascade of gene expression.
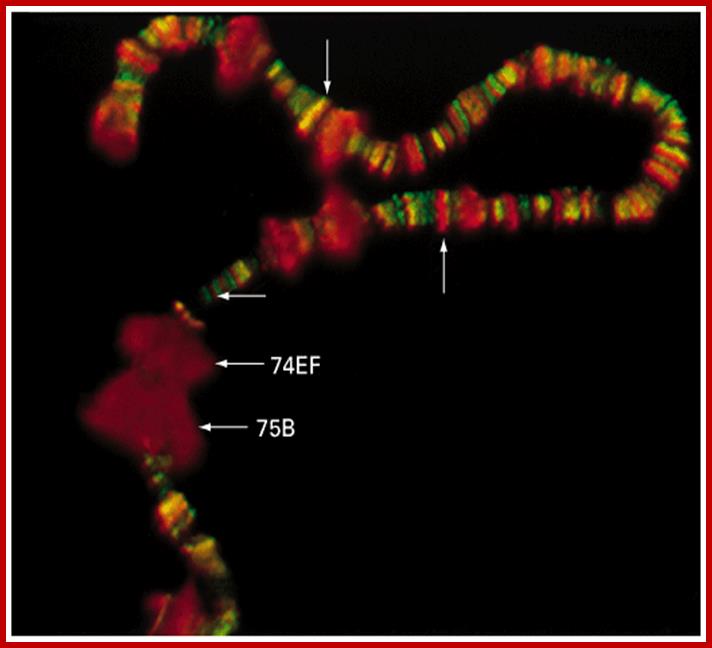
Chromosomal puffs at 75EF and 74B are visible
http://bioinfosu.okstate.edu

http://www.yourarticlelibrary.com
The polytene chromosome in its glory with 1080 duplicated strands show many very prominent chromosomal bands. Each of the bands represents a gene or group of genes. In some of the bands DNA loops out, free from associated structural proteins, 10kb to 50kbp size associated with transcription machinery producing transcripts required to be translated and some to be stored. The loci at which thousands DNA loops opened and expressed, looks like puffs (chromosomal puffs) when stained; some of the long loops, identified by Balbiani are called Balbiani rings. In between many chromosomal bands remain unexpressed. During transition from larva to pupa gene expression or puff formation and regression at different positions can be observed. Here again one observes chromosomal DNA at specific positions should be free from histone complexes to produce massive amount of transcripts.

Balbiani Rings: Electron micrograph of a Balbiani ring (A) and a schematic presentation of a transcription loop with growing RNP particles (B). The loop consists of three portions: an upstream region (a), the transcribed template (b), and a downstream region (c). In the electron micrograph, two segments of transcription loops have been indicated by arrows. The bar equals 1 µm. From- Daneholt (1992).

Specific loci containing a set of genes related are expressed in high order, thus one can see the Puff like regions. During formation stage, in salivary glands, chromosomes undergo repeated replication without separation to produce multistrands. This provides a scope for a gene to be present in 1080 copies; this provides an opportunity for transcription is massive scale.
Gene Expression Requires DNA of the Promoter either to be Free or to be loosened from the Grip of Histones and Repressor Proteins:
From the above description, for gene expression, the DNA in the chromatin has to be dissociated or freed from compacting histones and other associated proteins; for they block specific sites for the binding of activators and transcriptional complex. Chromosomes’ composite structure is made up of four histones. Histones constitute structural components of chromosomes as organized structural elements called nucleosomes. But chromosomes are also associated with thousands of nonhistones, which can be differentiated from histones from their very nature of being little acidic; not all of them. Most of them are involved in regulation of gene expression and DNA replication, DNA damage repair; and maximum number of nonhistones found on chromatin is found to be topoisomerases at the base of chromatin loops where the DNA is bound to scaffold proteins. Topoisomerase II has important function in removing super coiled DNA into relaxed forms either at replication stage or at transcriptional stage or whenever required; they can also induce super coiled structures and make it inactive. Can positive supercoiled DNA blocks transcriptional initiation? Why one finds such huge number of topoisomerases? Along with Topoisomerases another protein that is found in large amounts is a DNA binding protein called High mobility group {HMG); they are a family of proteins involved in binding and bending DNA and transcription. Another protein found at scaffold region is an 85KDa protein; they are like SMC proteins with ATP binding sites.
One notices the presence of gene activation and gene repression proteins all along the chromosomal threads, their loci depends on the specific DNA sequences, cell type and stage at which they are found. The number of such proteins involved in gene expression and repression may run into thousands. Delineation of it requires the expertise of functional genomic professionals. In this description, the regulation of gene expression is dealt in two different functional aspects but with inter-related structures and functions.
Bacterial genomic DNA which is also associated with proteins at their promoter regions, not so compacted as found in eukaryotic systems. The compacted DNA into chromatin often undergoes tight compaction and relaxation. The compacted regions can be observed by staining as dark bands.
The Role of Histones and Nonhistones in General:
It is very well documented that all cells in all tissues express certain set of genes for house-keeping functions, common to all cells in the body, they are called house keepings genes. Some are expressed in tissue specific manner; they are expressed using tissue specific factors. There are genes, whatever tissue or cell types, are exposed to certain signal molecules, and they induce signal specific gene expression. All other genes remain unexpressed or silent. The genes that are expressed have access to transcriptional apparatus and those not expressed or kept silent don’t have access to transcriptional proteins or the access is blocked.
In this context it is important to know which of the chromosomal proteins prevent transcription and which of them allow transcription in specific manner? To answer this question scientists have isolated DNA from an organism. They also isolated histones and nonhistones from different tissues. Very simple experiments (even high school students can understand) are designed to show which of the prime chromosomal proteins are involved in transcription and that too in tissue specific manner and which proteins block transcription.
First experiment:
DNA isolated from a system is same; common DNA for the experiment. Histones and nonhistones are isolated from specific tissues i.e. from Liver (L) and Brain (B).
In one experiment non histones from liver and brain tissue are added separately to the common DNA and then histones were added and allowed to express; experiments showed nonhistones from liver expressed liver specific genes and brain specific nonhistones expressed brain specific genes. Histones could not prevent nonhistones’ mediated expression. Tissue specific expression is due to specific nonhistones.
In the second set of experiments histones were added first to the DNA, and then non histones from brain and liver added separately, results showed gene expression failed, for the histones binding to DNA blocked access to non-histones. Though this experiment is simple yet its result showed nonhistones find their promoter elements and initiated transcription, histone cannot block when nonhistones already bound, but when histones are bound nonhistones cannot act. Identification and characterization of those nonhistones involved in gene expression is fascinating.
Second experiment:
Isolate a specific DNA segment, say 5s rDNA, add histone first then add required components such as IIIA, IIIC, IIIB and RNAP III; result no transcription. Instead add IIIA first then add histones and then other components; transcription takes place. This shows the binding of IIIA to specific sequence is a gene identity factor; it does not allow histones to inhibit. Once IIIA binds in sequence specific manner; when it binds to its sequence, it recruits other components to initiate transcription even in the presence of histones; so histones cannot prevent transcription once sequence identity factor binds; i.e. ‘nucleating’ factor binds. Can nucleating factors perpetuate through cell lineages? Yes, in bacteria they do and there no doubts that eukaryotes have this facility, though not completely elucidated.
Inference:
In eukaryotes the promoter elements have more complex combination of sequence boxes to be recognized by a combination of factors. The transcriptional activators bind to different elements located at different positions away from the promoter and START site. So also transcriptional repressors, they do so by binding to specific sites identified by sequences. So activators activate specific gene transcription and repressors repress specific gene expression. Repressed genes can be induced to express and active genes can repressed when required. There are others called silencers and insulators, which have their own specific roles to play.
The regulator proteins of one kind are activators, co-activators, mediator complexes and transcriptional complexes. The others are repressors, corepressor and their associated proteins. They bind to different structural elements and activate the gene expression or gene repression in specific. Activators first seek specific promoter sequences and bind if the region is free from histones, then co activators, Mediator complexes join, the latter don’t bind to DNA. Repressors are specific to specific genes, so also specific silencers. In some cases the repression is not just few loci but the whole chromosomal genes are totally inactivated, in an extreme case, but it is prevalent in higher systems for example inactivation of human X chromosome. In many of the chromosomes certain regions of chromosomes are repressed such regions are called heterochromatin.
It is important to know repression can be due to specific repressor binding and preventing the access to transcriptional machinery; or the repressor remains in place until it is induced to change either to become activator or they get released from the DNA. There is another mode of repression that is heterochromatization. One such state is telomeric region and pericentromeric region, where the chromosomal region has constitutive heterochromatin. There are other heterochromatin blocks in euchromatin such and they found to change the loci from one tissue to the other; they are called facultative heterochromatin; it is the facultative heterochromatin that blocks some blocks of genes. In some cases certain gene expression is blocked by localized heterochromatization. There are many such examples due such abnormal heterochromatization leads to epigenetic expression or repression of genes that are unrelated to normal gene expression and cause diseases. Histones should not be considered as sole repressor proteins, but they are fundamental building blocks of chromatin where DNA is strengthened as the fiber and prevent DNA from shearing and breakdown. They provide remarkable strength for the DNA 10A^o thick to remain unsheared during cell cycle and gene expression. Another very important structural protein that reinforces and sustains the rigors of chromosomal changes during replication, recombination and DNA breakage and repair is that of scaffold protein; it also sustains several modes of chromatin changes such as relaxation and differential compaction.
Chromosome Remodeling Protein Complexes and their Function:
Important Gene Regulation Factors:
In eukaryotes, the DNA, for that matter any organism, such as bacteria or viruses (some viruses contain RNA as the genome), that has encoded message, contains promoter elements with more complex combination of sequence boxes to be recognized by specific factors. DNA is associated with histones and nonhistones and organized into chromatin thread. This, when it is in relaxed i.e 11-to 30nm state the chromatin is open and transcription complex can assemble; but if it is compacted, the transcriptional factors cannot access the promoter, so no transcription. Chromatin in relaxed state is basic 11-30nm chromonemal thread where nucleosomes are bound together by Histone1. This thread in 30nm state, where the chromonemal thread loops out and its base is bound to scaffold region. This is subjected to activation or repression of genes. It is the same thread gets compacted differentially into heterochromatin and can be relaxed as euchromatin. Repression is not always due to heterochromatization; but it is also due to the binding of specific repressor complexes associated with 30-110nm structure. Binding of one factor induce and recruit other accessory factors either for activation or repression. In order to understand the complexity of activation and repression one has to look deep into complex components of proteins and their role.
Chromatin Activators and Repressors:
Activators bind to specific elements located at different sites upstream of the START site. So also transcriptional repressors bind to specific sites; they are identified by sequences.
The structural and functional features of both gene activators and repressors have specific motifs and domains, each have specific functions in gene regulation. In spite of the variety of components involved in regulating gene expression, the overall mechanism, from yeast to man to plants, is more or less similar. Why? For expression of genes chromatin in specific regions has to open and provide access to transcriptional factors and RNA polymerase for binding and initiate transcription.
Basic structural components of chromatin are histones and DNA; in addition a large number of non-histones are also found associated with basic histone DNA threads. If one observes chromatin in vivo, its surface shows the chromatin is studded with whole lot proteins not just histones. Probably every gene or gene loci are already associated with specific proteins; identity of them and their location is important.
In general, what is now known today that it is histone components bound to DNA and their structural modifications that make the chromatin to be in inactive or active? Though histone octamer enwrapped around by DNA and nucleosomes are compacted by H1, yet nucleosomal histone tails of ~42nm long in comparison to ~65bp long linker DNA interact with one another and their modifications perform most enticing functions. Modification of histone tails and additional components added to such tails make the chromatin compact or loosen. Even DNA CpG modifications also contribute to its compactness. Understanding of such modification is very important in understanding chromosomal remodeling and regulation of gene activity. At chromosomal level histones and nonhistones perform functions as to the cell requirements. The crux of the problem is that chromatin DNA should be free from histones so that activator or repressor factors can bind. If chromatin is compacted DNA won’t be free for the binding of these regulator proteins. The 30nm chromonemal loops which are bound to scaffold protein (300nm structures) should provide access to factors for the binding. But this chromatin is compacted differentially into tight compaction and loose compaction, the former is called heterochromatin and the latter is called euchromatin. Even euchromatin is not free for the binding of factors for H1 is bound to linker DNA and compacted to some extent. What is interesting is that the H1 binding and the positions of nu bodies are dynamic, in the sense they assemble and disassemble and the DNA in euchromatin is very often made free and close in short period of time. Is this time period is enough for the assembly of factors to bind and execute their activity? Another important aspect of the chromatin is that whether or not all those nonhistones bound before the replication of chromatin DNA remain associated when the daughter DNAs or after DNA replication, do they reassociate afresh?; if so this has to happen even before the chromatin thread is formed.
It is known that cells perpetuate their characters to their lineage of cells, but the pluripotent cells undergo differentiation with every cell division. Maintenance of cells structure and functional features in their lineage of cells should contain all the regulatory factors bound to their respective positions. There is a lot of bias among the scientist in interpreting this perpetuation. Perpetuation of old histones and complementing them with new histones is known. Perpetuation of DNA methylation is also established. Similarly modified old Histones remain associated and evenly distributed among the newly formed DNA strands. Perpetuation of repressors and activators in prokaryotes is established. Similarly perpetuation of nonhistones is not unthinkable. Take for example, the GAL4 protein as repressor remains bound to chromatin for many generations of yeast cells. After its activation the same is also perpetuated in dividing cells.
Histones and histone modifications and their effects on chromatin:
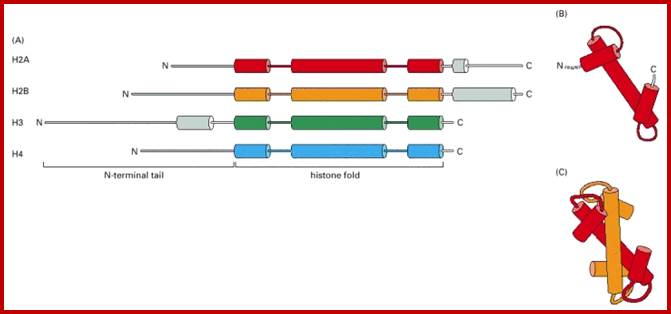
Histone fold- most of the histones contain a central helix domain with smaller helices on either side of the main helices, and also contain N terminal tails with specific amino acid sequences, but H2A in addition to NH2 tail contains C terminal tail too. Histone tails with specific a.a sequence get modified-this feature provides what is called as Histone code.
Chromatin II, histone modifications: http://bricker.tcnj.edu/
Epigenetic regulation of key vascular genes and growth factors- Histones are subject to post-transcriptional modifications, which occur in histone tails. The best-known post-transcriptional modifications (acetylation and methylation) are shown. The number under each amino acid represents its position. Greek ‘Epi' means ‘over, on-top or above', therefore Epigenetics refers to something above genetics itself. Epigentic marks are not in DNA sequence itself but on top of them, as chemical additions on DNA stretch (DNA methylation, Figure 2) or on those proteins in which the DNA is wrapped around (Histones, Figure 1). These modifications act as switches turning gene expression on or off. Functional epigenome is necessary for health of a cell or organism. Epigenome is likely an easier target for therapeutic modifications when compared with genome itself; http://cardiovascres.oxfordjournals.org/
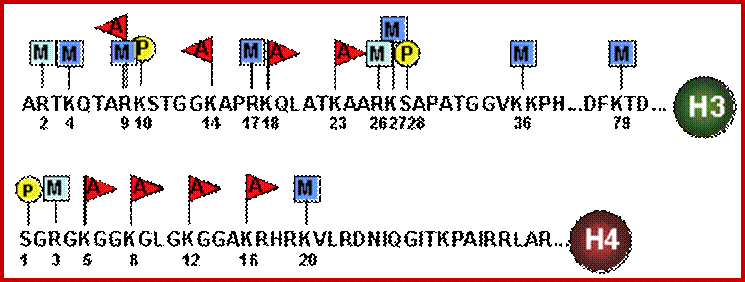
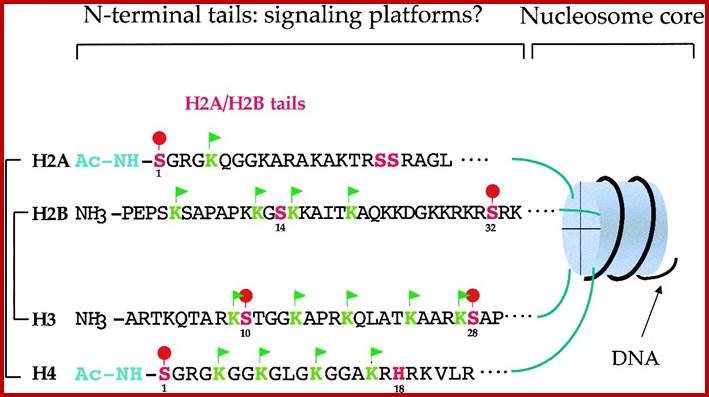
N terminal tails of H3 and H4, their modifications play very important role in chromatin condensation or relaxation; for that matter activation of genes and repression of genes. Position of specific amino acids is indicated for specific kind of modifications; such as methylation, acetylation and phosphorylation. Some amino acids are get modified either by acetylation or methylation. If they acetylated they can be removed by deacetylases and if they are methylated they are demethylated by demethylase.
Arginine is preferred for methylation; but R can also be acetylated. But K in certain positions as shown in the figure is mostly acetylated and in some positions it can also be methylated. Rarely it can be ubiquitylated. Serine is the most preferred site for phosphorylation. It is important to know the site of amino acids in the tail and its modification provides what is called histone code. Such modifications provide the information for the binding of other proteins.
DNA in eukaryotes is packaged into nucleosomes which consist of DNA wrapped around histone proteins. Covalent modification of histones plays a critical regulatory role in controlling transcription, replication and repair. Different histone modifications are recognized by different protein modules found in regulatory complexes with different, even antagonistic functions; acetylated sequences are recognized by Bromodomain proteins and Methylated sequences are recognized by Chromodomain proteins. Positions of acetylation, methylation, phosphorylation and ubiquitination (SUMOylating) are shown below.
The chromodomain helicase DNA binding protein 1 (CHD1) is well known for its remodeling activity in the maintenance of stemness. It also has main function in recognizing a substrate of transcription regulatory histone acetylation complex SAGA. CHD1 has been suggested to act as a molecular adaptor, which bring several epigenetic complexes together [29]. In ESCs, this adaptor has been suggested to be indispensable for the maintenance of pluripotent chromatin state where it is highly expressed when compared to differentiated cells. After knockdown of the CHD1 with RNAi, the pattern of diffuse ESCs heterochromatin disappears showing a higher amount of heterochromatin. In turn, CHD1 knockdown fibroblasts reprogrammed less efficiently [30]. The nature of CHD1 in pluripotent cells specifies that it can prevent the formation of heterochromatin foci [30]. CHD1 has also been reported to be one of the genes that activate Oct4, Sox2 and Nanog; http://www.intechopen.com/
Histone modifications have been associated with either 'active' or 'inactive' chromatin states, as well as with particular cellular processes, including mitosis, spermatogenesis and DNA repair. Some modifications, such as histone lysine methylation, are known to recruit specific binding proteins (for example, HP1 to methylated histone H3 lysine 9 and PRC1 to methylated histone H3 lysine 27, whereas acetylation at various residues is believed to have a more structural role, making the nucleosome structure 'looser' and more accessible to transcription factors. Several synergistic and antagonistic interactions have been described between different histone modifications. On the basis of these observations, it has been proposed that patterns of post-translational modification form a combinatorial 'histone code’. However, the degree of interdependence between different histone modifications, and the various distinct chromatin states they define (individually or in combination), the state of chromatin; this feature is still not entirely understood


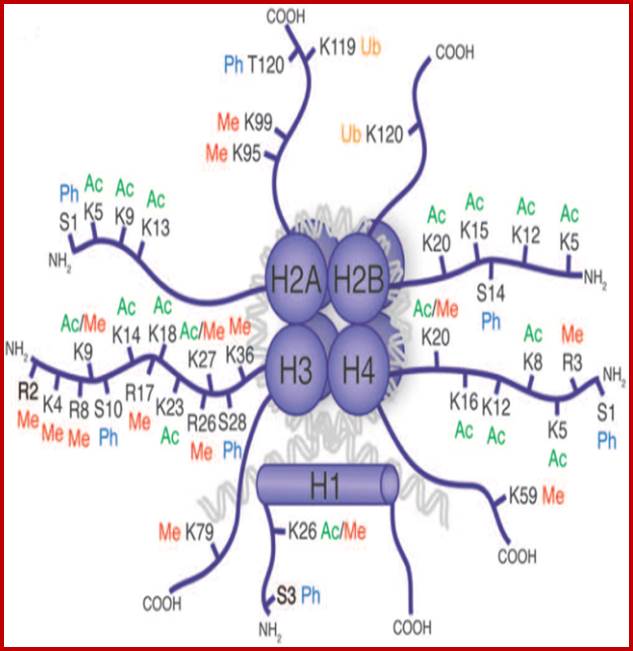
H1 = 208 (222) aa, has central globular body with N and C terminal tails. H1 composition varies among the species. It has a 5 R s and 53 Ks and 5 D & Es.
H2A = 129 aa, it contains both C (39 aa) and N terminal tail- N S G R G K Q G G K A R A K A K T R S S R A G L, with 12 R, 14K and 7 D/ Es.
H2B = 126(125) aa, it has 34 N-terminal tail with 7R, 21K and 10 D and Es-
P E P S K S A P A P K K G S K K A I T K A Q K K D G K K R K R S R K.
H3 = 136 (135) aa, its N tail 32 contains 18R, 13K and 11 D n Es-.
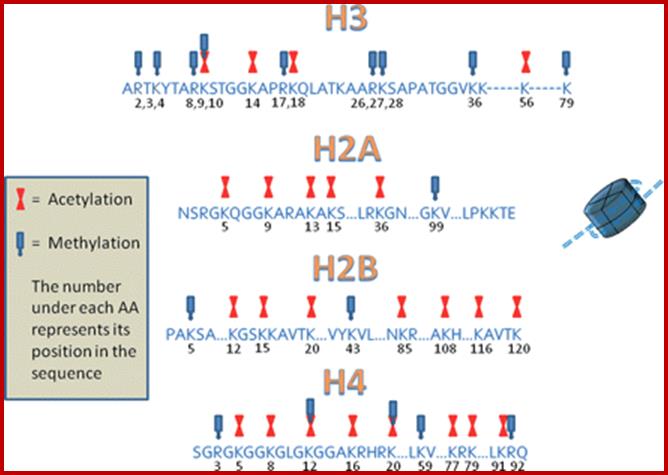
-A R T K Q T A R K S T G G K A P R K Q L A T K A A R K S A P A T G G V K K; where K9 and K14 are acetylated in euchromatin, even S9 can be phosphorylated.
H4 = 103 (102) aa, N-tail 32 contains 14R, 11K and 7 D n Es-n-S G R G K G G K G L G K G G A K R H R K V L R D where R3 can be acetylated in euchromatin.
![]()
Structural features of Lysine and Arginine: http://www.dls.ym.edu.tw/

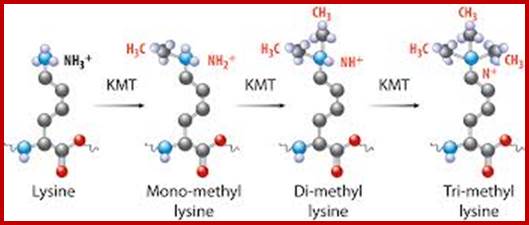
Chromatin-Modifying Enzymes- The diversity of chemical states obtained by selective and sequential methylation of lysine and arginine residues within proteins, as catalyzed by the histone methyltransferase class of enzymes. Copeland et al (2012) Targeting Genetic Alterations in Protein Methyltransferases for Personalized Cancer Therapeutics. http://www.epizyme.com/epigenetics.

Lysine acetylation and deacetylation by specific acetylases and deacetylases
Histone tail modifications and their functions:
|
Histones |
Site/ position |
Modifica- tion |
Effect |
|
H3 |
K4 |
CH3 |
Activation/ Recruit H2A macro |
|
H3 |
K9 |
Acetyl |
Activation |
|
H3 |
K9 |
3CH3 |
DNA methylation/ Mecp2 binds |
|
H3 |
K14 |
Acetyl |
Activation |
|
H3 |
K9 |
CH3 |
HP1 binds |
|
H3 |
S10 |
PO4 |
activation |
|
H3 |
K79 |
Acetyl |
Telomere silencing |
|
H4 |
R9 |
CH3 |
Inactivation |
|
H4 |
K5 |
Acetyl |
activation |
|
H4 |
K5K12K16 |
Acetyl |
activation |
|
H4 |
K20 |
3CH3 |
Xist RNA, X chromo methylation |
|
H4 |
K20 |
|
|
|
H4 |
S 1 |
P-lation |
Chromo compaction |
|
H2A |
K119 |
ubiquitin |
Xist, SiRNA |
|
|
|
|
|
|
|
|
|
|
Modification of H3 and H4 tails has important effect on chromatin compaction or relaxation. The K can be acetylated or it can be methylated, similarly R can be methylated and serine can be phosphorylated. These modifications can be reversed i.e. deacetylation, demethylation and dephosphorylation. Specific modifications at specific sites have tremendous effects on chromatin activation (loosening) and chromatin inactivation (compaction).
acK = Acetylation to K (lysine,
meR = methylation to R (Arginine),
meK = methylation to K,
pS = phophorylation to Serine,
pT = phosphorylation to Threonine
uk = ubiquitination to K,,
The order of amino acids in the H3 and H4 tails play significant role in chromosomal activation or inactivation i.e activation of a gene or suppression of a gene. David Allis proposed that histones N-terminal amino acid sequences provide encoded information, what is called Histone code, where certain modifications evoke certain chromatin based functions, a combination of modifications evoke specific biological functions.

It is not only Lysine and Arginine gets modified, even DNA at specific dimers of CpG cytosine is added with methyl group.
Role of histones:
Organization of Nucleosomal thread, provide strength and stability to DNA.
Packing nucleosomes with H1 lead to chromonemal thread formation.
Histones with non-histones produce chromatin.
Modified histones mostly with CH3 can lead to compaction and gene silencing,
Modified Histones with acetylation relax chromatin and genes can be activated,
Modifications of Histone tails in the region of promoter elements have a role in heterochromatization and relaxation of the same. Histones perse don’t bind to specific regulatory elements of genes; but nucleosomal threads provide stability and strength to DNA. However association of histones with DNA is random and there is every possibility, they may cover regulatory sequence of genes.
Non-histones and their role:
There are more than 2000 nonhistones proteins,
Most of them are slightly acidic or neutral,
Topoisomerase II is found in large quantity,
HMG family of proteins is another found in large amounts,
Another protein is 85 KD (?) whose function is not clear, a scaffold protein?
Chromatin is associated with large number transcriptional complexes-transcriptional regulators-activators, co activators, repressors, and corepressor, silencers and insulators, which are all grouped as nonhistones.
Their quantity and quality of protein varies from one tissue type to the other.
Histone modifying enzymes and enzyme complexes:
|
Complex |
System |
acronym |
function |
|
|
SAGA |
Yeast |
HAT |
Histone acetylation |
|
|
PCAF |
Homo |
HAT |
Histone acetylation |
|
|
STAGA |
Hu |
HAT |
Histone acetylation |
|
|
NuAA |
Yeast |
HAT |
Histone acetylation |
|
|
HDAc1 |
Hu |
HDAc |
Histone deacetylation |
|
|
SMRT/N-CoR |
Hu |
HDAc |
Histone deacetylation |
|
|
RPD3 |
Yeast |
HDAc |
Histone deacetylation |
|
|
SUV39H1 |
Hu |
HMT |
Histone methylation |
|
|
SET1 |
Yeast |
HMT |
Histone methylation |
|
|
|
|
DMT |
DNA methylation |
|
|
|
|
|
|
|
ATP-dependent chromatin modifying enzyme complexes:
|
Complex |
Organism |
function |
|
RSC |
Yeast |
Restructure |
|
SWI/SNF |
Yeast, fly, Hu |
loosening |
|
NuRF |
Fly,Hu |
Nu restructuring |
|
CHRAC |
Fly, Hu |
Chromosome associate |
|
ACF |
Fly |
ATP- Utilizing |
|
ISW1/2 |
Yeast |
Imitation of SWI |
|
RSF |
Hu |
Restructuring |
|
M1-2/NuRD |
Hu |
Nucleosome R deacetylase |
Chromatin remodeling complexes:
SWI/SNF = 11 subunits, 2MD, has helicase activity, slide nucleosomes, ATP mediated chromatin remodeling factor, involved in regulation of cell cycle, cell differentiation, transcriptional regulation, cancer inducing; involved in immunity, acts at 120 loci (yeast), 150 complexes per cell,
SAGA/PCAF = number of subunits~20, 1.8 MD, found in EK (eukaryotes) included in GcN5/ADA, involved in acetylation facilitating transcription,
SAGA = Spt-Ada-Gcn5-acetyltransferase complex
NURF = 4 subunits -0.5MD, found in Droso, involved in chromatin remodeling, consisting of subunits 215, 140, 55 and 38KD subunits. Nucleosome stimulated ATPase (Nucleosome Remodeling Factors).
RSC = 15 subunits 3MD involved in restructuring complexes of chromatin, acts at 700 loci
CHRAC/CAF1= subunit2, 0.5Md, helps in H3/$ binding to DNA, Chromosome Accessibility Complex).
ACF = ATP dependent chromatin assembly and remodeling Factor.
Trithorax= huge complex involved in activation of some HOX genes during development,
PCG= Polycomb group complex involved in repression of some HOX genes during development,
SWI = It is a yeast mating switch complex and contain ATPase units.
CRC = Chromosome remodeling complexes General.
SNF- Sucrose non Fermentor associated with SWI.
BAH- components are proteins associated with bromodomain binding factors. Bromodomain consists of 110 a.a sequence which binds to modified histone tails mostly acetylated and recruits more HATS.
ISWi- it is an imitation of SWI
RPDs-Some of the RNAP associated transcription complexes are called RPDs.
RPD3- is a histone deacetylase.
NURD = Nucleosome Remodeling Deacetylase,
CAF = Chromosome associated factor
The remodeling complexes are grouped-
1. SWI/SNF complexes (include ATPases sw2/snf2, RSC, Brahma BRM (Brahma related protein) BRG1-Brahma related gene 1.
2. ISWI- imitation switch components, ACFs ATP utilizing chromatin assemble and remodeling factors, CHRAC-chromatin accessibility complex. NURF- Nucleosome remodeling factor and NSF-Nucleosome remodeling and spacing factor.
3. Mi2 complexes –they include human NURD also contain HDAC1 and HDAC2.
4. HP- many HPs chromodomain proteins.
It is known some these factors are associated or bound to transcriptional activators or repressors. Having ATPases factors use ATP energy they distort binding of DNA to Histone and push the histone complexes along the DNA; they free the DNA from nucleosomal structure, so a segment of DNA is left free for the assembly of Transcription factors and their associated components. Methylation of certain histone tails recruits HP1 proteins which compacts chromatin to condensed heterochromatic state.
Note: ACF= ATP-utilizing chromatin assembly and remodeling factor; CHRAC = chromatin accessibility complex; HDAC= Histone deacetylases; HMT= histone methyl transferase; ISWI-2= imitation of switch protein; Mi2NURD= Nucleosome remodeling complex; NUA4= Nucleosome acetyl transferase H4; NuRF = Nucleosome remodeling factor; PCAF= p300/CBP associated factor; RSC= Remodel structure of chromatin; RCF= Remodeling and spacing factor; SAGA= Spt-Ada-GCN5-acetyl transferase; SET= SET domain containing protein; SMRT/NCOR= silencing mediator of retinal and thyroid receptor/nuclear receptor co repressor; STAGA= Spt3-TAFII-31-GCN5L acetylase; SUV39H1= Suppressor of variegation 3-9homologue-f; SWI/SNF switching defective/sucrose Non Fermentor.
Chromatin is a complex of DNA-histones as nucleosomal thread superimposed by nonhistones. Such a thread is often exists, in certain regions of the chromosomes, as condensed and relaxed threads. It is believed that relaxed region is more open for transcription than the condensed ones. Remodeling means the chromatin condensed to be decondensed; and decondensed chromatin to be condensed; it is done by chromatin remodeling proteins.
This remodeling of chromatin is performed by certain multi-subunit protein complexes that are associated with chromatin. It is also important to remember that there are several thousands of proteins, called site specific identifier proteins, bound to chromosomal DNA they form part and parcel of chromatin thread. Some such bound proteins are perpetuated and some may be added or removed with time and state of the cell. Multi subunit complexes which are ATP dependent provide fluidity to chromatin. The chromatin is also associated with chromosome remodeling multi subunit protein complexes. Most of them ATP driven, so they remodel the nucleosomes to move from one site downstream so a region containing promoter is made available for regulatory proteins to bind and act.
One such complex is SWI/SNF, they were discovered in yeast and they make HO genes to express. (SWI means switching mating defective). And SNF means Sucrose Non Fermentor. The SWI/SNF complex is 110KD consists of 11 subunits. It is able to express 3% of the yeast genes.
Another complex called RSC (Remodel the Structure of Chromosome) is 100 times more abundant than SWI/SNF and they share two subunits of SWI/SNF. RSC contain homologs of SWI/SNF, called as Sth1, they are called as swi2/snf2 in SWI/SNF. SWI/SNF are ATPases and two of the subunits of RSC have bromodomain so they bind to acetylated Histone tails.
Chromatin remodeling complexes and mol.wt and subunits:
|
Complex |
System |
ATPase |
Mass |
subunits |
|
|
SWI/SNF/family
|
|
|
|
|
|
|
SWI/SNF |
S.c |
SWI2/SNF2 |
2 |
11 |
Chromo loosening |
|
RSC |
Sc |
Sth1 |
1 |
15 |
Remodeling complex |
|
|
|
|
|
|
|
|
Brahma |
D,m |
Brahma |
2 |
no |
Acetylase |
|
hSWI/SNF |
Hu |
hBRM |
2 |
>10 |
remodeling |
|
hSWI/SNF |
Hu |
BRG1 |
2 |
>10 |
|
|
NuRD |
Hu |
HD4 |
1.5 |
18 |
Nuclosome Deacetylase |
|
ISWI family |
|
|
|
|
|
|
ISWI1 |
S.c |
hISWI1 |
0.4 |
4 |
|
|
ISWI2 |
S.c |
hISWI2 |
0.3 |
2 |
|
|
NURF |
Dm |
hISWI |
0.5 |
4 |
Nucleosome remodelng |
|
CHRAC |
Dm |
hISWI |
0.7 |
5 |
|
|
ACF |
Dm |
hISWI |
0.2 |
4 |
|
|
RSF |
HuI |
hISWI |
0.5 |
2 |
|
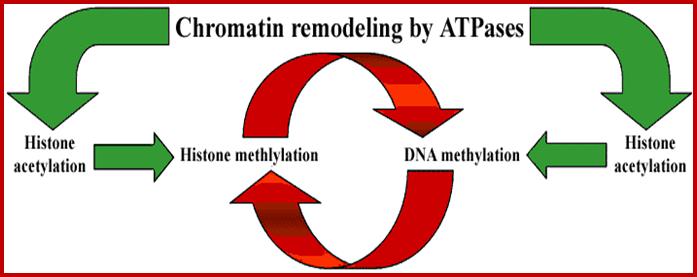
Remodeling Protein domains:
|
Domain |
Target |
Function |
|
Bromo |
Acetylated Histones |
Transcription, Repair, Replication, Condensation |
|
Chromo |
CH3H3K27 CH3H3K36 |
Silencing, Activation |
|
OHD/Bromo |
CH3H3K4 |
Activation |
|
TUDOR |
CH3H4K20 CH3H3K4 |
DNA repair/ transcription |
|
PWWP |
CH# Hi/DNA |
? |
|
MBT |
Ch3H4K20 |
DNA repair |
|
|
|
|
|
|
|
|
Chromatin activating factors: GCN4, GCN5, GCN5L ADA, SAGA, SWI, SNF, NuRF, CAF1, NAP1, CRC, NUA4HAT,Trithorax, hTAFII250, GRIP, TIP60complex, RSC, CPB/p300, PCAF, Src1, ACTR and ELP3
Chromatin inactivating factors: NURD, SIN3 family, MBP, MBD1/2/3/4, MeCp2, Histone deacetylases 1, 2, 6, 7, 9, 10, 38 ,45, Histone methylases, HP1 family, RAP, RIF, AIR1, 2, 3 and 4, SUMO l, SW6, Chp2, Clr3 (Chromodomain).
Post-translational modifications of the core histone tails that stick out from the nucleosomes have been directly linked to the regulation of chromatin structure, a concept known as the histone code. Modifications of the core histones include acetylation, methylation, ubiquitination and phosphorylation. Such modifications alter the interactions of histones with DNA and the recruitment of chromatin associated proteins. The best characterized histone modifications are acetylation and methylation. Acetylation and methylation of histone tails are carried out by histone acetyl transferases and histone methyl transferases. Acetylation is primarily associated with active gene expression, but methylation at different positions perform silencing and activation.
Histone acetylation results in the relaxation of the basic chromatin structure through increased charge repulsion and by serving as binding sites for protein complexes of chromatin-modifying and transcriptional activators. Histone methylation can be found in both heterochromatin and euchromatin too. H3 methylation of K4 is found in activated gene promoters. Trimethylation of histone H3 on lysine residue 9, (H3K9me3), is bound by heterochromatin protein 1 (HP1), which results in chromatin compaction and heterochromatin formation. This pattern of histone modifications causes gene silencing or activation can be inherited to their daughter cells, a phenomenon called epigenetics or it can happen during the course of life span.
Chromosomal State during Gene expression:
Regulation of gene expression in eukaryotes is more complex and intrinsic. The genome is organized into a nucleoprotein complex of different orders. Chromosomes bear genes of different types such as coding for proteins, rRNA, tRNA and other Nc RNAs such as RNAi and few others. They are expressed differentially during development and also in tissue specific manner during and after development. Even after development genes are expressed in tissues in response a variety of signals.
Chromosomes go through condensation and decondensation differentially during cell cycle and even after cell cycle. Chromosomes undergo structural changes, from relaxed state at interphase or G˚ to highly condensed state at metaphase. At interphase substantial number of genes is expressed in tissue specific manner. At metaphase the chromosome is condensed to such an extent all genes are shut off. Once the cell derivatives differentiate and develop into specific cell types, chromosomes in relaxed form found attached to the inner surface of the nuclear membrane protein matrix; the attachment is to heterochromatic loci. Most of it is in the MARs and SARs region of the DNA (300-1000bp long) that contain repeat ~AT rich sequences. The Scaffold Associated Region (SAR) binding polypeptide SATB1 is found associated with MARs. The nucleus consists of pervasive nuclear matrix proteins (Nuclear matrix is analogous to the cell cytoskeleton), whose composition and nature is not yet clear. The relaxed euchromatin region is accessible to transcriptional complex, it does not mean that all those genes present in euchromatin region are expressed, it is not so.
It is logical to expect that chromosomal loci where gene to be expressed requires unwinding of nucleosomal 30nm structures into relaxed form and at least some part of the DNA of the said gene is to be free from histones for the binding of regulator proteins and its related factors.
Whether regions that are active in gene expression or not can be tested by DNase1 treatment or micrococcal nuclease, which on partial digestion of the DNA wherever it is free DNA is digested completely and wherever nucleosomes are found, only the linker DNA is digested. Nucleosome bound regions show ladders and nu free regions are completely digested.
DNase I hypersensitivity, and computational approaches have been employed to generate maps of histone modifications, open chromatin, nucleosome positioning, and transcription factor binding regions of mammalian genomes. Given the importance of non‐promoter elements in gene regulation and the recent explosion in the number of studies devoted to them, many scholars have focused on these elements to get insights on gene regulation.




Chromatin state can be observed when micrococcal nuclease is used to digest chromatin and digested pattern can be observed on gel electrophoretic image. So the DNA of a gene is in active state should be free from histones, and mostly it is of promoter regions; it is the promoter region where Transcriptional apparatus and its associated factors bind upstream of InR.
Chromatin remodeling at the site of gene expression:
David Allis concept of Histone code provides an input where active chromatin, means any gene(s) in that region are expressed or expressing. Such chromatin shows its H3 tail is acetylated at K9, K14 and H4 acetylated at K5 and its H3-K4 and H4 R3 are methylated (in general). But condensed chromatin i.e. inactive is associated with H4-K12 acetylated, and H3’s K9 is methylated. Nucleosomes are also disposed for phosphorylation at Serine10 and 28. The combination of modifications at specific amino acids in the N-tails H3 and H4 have intrinsic ability to change the shape of chromatin to open for transcription or close to transcription at specific loci or sites. This state of histone modified chromatin varies from loci to loci.
Histone modifications during early cell division, cell determination and differentiation provide that epigenetic markings assisted by the binding of specific factors to specific sites in the region provides the identity of the gene or genes to be expressed or not to be expressed in specific tissues or in specific environment or inducer or repressor signaling. This epigenetic inheritance of histones during determination and differentiation of cells is yet be discerned beyond doubt. It also provides markers for the association of gene specific or tissue specific factors or both, thus provides the inheritable constitutional input for the next generation of cells or tissues, which can respond to various inputs. To give a simple example of yeast GAL genes regulation; GAL1, GAL7 and GAL 10 are located nearby on the chromosome 11 and their gene products are involved in utilization of Galactose in the absence of Glucose. The promoter elements of the said genes located at specific loci are bound by GAL4. The GAL4 binding remains at the location for any number of generations of cell lineages and the said genes remain repressed.
GAL4 is actually an activator, but GAL 80 by binding to GAL4 suppresses GAL4’ activators function. This is an eukaryotic unicellular system; this structure and functional features may apply to all multicellular system. When GAL4 is activated by release of GAL80, it recruits SAGA complex which facilitates transcription by remodeling nucleosomal structure and facilitate transcription.

Fig: A model for SAGA functioning as a co activator for Gal4 by facilitating TATA-binding-protein (TBP) binding to the TATA box of the GAL1 gene. (1) Under noninducing conditions, Gal4 is bound to the UASG via its DNA-binding domain (DB), and the Gal4 activation domain (AD) is blocked by Gal80. (2) After the addition of galactose, the Gal3 inducer is activated and alters the Gal80–Gal4 complex such that the Gal4 activation domain is no longer blocked by Gal80. This change allows the Gal4 activation domain to recruit SAGA to UASG. The presence of the Gal3 protein at the promoter is suggested by the formation of a Gal3–Gal4–Gal80 complex in vitro and in vivo (Chasman and Kornberg 1990; Leuther and Johnston 1992; Parthun and Jaehning 1992; Platt and Reece 1998; Sil et al. 1999). However, other data have suggested that Gal3 is cytoplasmically localized (Peng and Hopper 2000). (3) Once recruited to the promoter, SAGA, mainly via an Spt3-–TBP interaction, recruits TBP to the TATA box to allow transcriptional initiation; Nucleosomes are not shown.
It also suggests that cell types generated during development contain specific factors associated with chromatin, which on stimulus can be activate gene expression. Epigenetic is the hall mark of gene regulation in eukaryotic (EuK) and prokaryotic (PK) systems. In PK specific operons are repressed and some operons are expressed, because of the binding of repressors and activators respectively and perpetuate the same for many generations. It is very important to remember that majority of the genes are expressed as housekeeping genes, which are required as sine quo non components.
Dynamic Histone Acetyl Transferases / Deacetylases and Methylases/demethylases and Ubiquitinases:
HATs: Histone Acetylases:
In avian and mammalian cells,
transcriptionally active chromatin regions have core histones undergoing high
rates of acetylation and deacetylation, while in repressed chromatin regions
the rate of reversible acetylation and methylation is slow. Histone acetylation
is a dynamic process. Transcriptionally active chromatin has core histones that
are rapidly hyper acetylated (t1/2 =5 to 12 min for monoacetylated H4) and
rapidly deacetylated (t1/2 = 3 to 7 min). A second population is acetylated
(t1/2 = 200-300 min for monoacetylated H4) and deacetylated at a slower rate
(t1/2 = 30 min for monoacetylated H4).
Histone acetylation manipulates higher order chromatin and nucleosome
structure. The tails of H3 and H4 are important in fiber-fiber interactions,
suggesting that acetylation of the H3 and H4 tails will prevent chromatin
fibers from interacting (repulsion) with each other. Also, histone acetylation
has a profound effect on the solubility of chromatin in 150 mM NaCl or 3 mM
MgCl2. Acetylation of the core histones destabilizes histone-DNA
contacts and has a role in maintaining the unfolded structure of nucleosomes.
Thus, dynamic histone acetylation confers positional information for gene
expression; this takes place in the chromatin of 300nm where DNA loops of 30nm
are found.
Dr. Dave Allis' group was first to purify a nuclear histone acetyl transferase (HAT A) and to clone its cDNA (Tetrahymena nuclear HAT p55). HAT p55 was found to be homologous to yeast GCN5, a transcriptional adaptor/co activator with HAT activity. While GCN4 acts as a gene activator the GCN5 acts as components of co activator responsible for acetylation. GCN4 as a pioneer factor binds DNA in sequence specific manner (promoter). This pivotal discovery revealed how HATs were directed to specific chromatin regions for transcriptional activation.
HATs are components of multisubunit transcriptional co activators. It has been
found that most of the genes expressed are found in chromatin loci where their
specific histone tails are acetylated. And those genes remained suppressed or
not activated are found to have methylated or deacetylated or the combination
of both. This need not be the case in situations.
Vincent Allfrey (in 1960s) discovered histone acetylation and deacetylation involved in transcriptional activation and inactivation in EuK (eukaryotic) systems. In 1990 HAT and HDATs proteins involved were identified and they found to act at specific positions of the Histone tails and some of them are well characterized.
All HATs use acetyl-coA as group donors. As there a large umber of them they grouped as five HAT families.
GNAT family: Gcn5 related N-acetyltransferase; GCN5 was first found in
Yeast, it is well characterized; its related members are Gen5L, and PCAF, p300/CBP –and its associated factors; p300 and CBP means cAMP response element binding protein, they are homologous to trans-activational factors. Hat1 is found in cytoplasm and acetylate histones before they are transported into the nucleus.
MYST family: named after founding members- Moz, Ybf2/Sas3, Sas2 and Tip60.
P300/CBP family: Very common, specially found to operate in many steroid activated gene regulation.
TAF Family; they are associated with TBP protein in RNA pol assembly complex. TAF1 (also called earlier as TAFII 250) is the largest protein of TFIID complex that binds as the first component in the assembly of PIC.
The SRC family: steroid receptor co activators.
They have a variety o functions in transcriptional activation and also implicated in regulation of cell cycle and transcriptional regulation.
Most of the HATs act as multiple subunit complexes of 10-20 subunits, they form complexes such as SAGA (spt/Ada/Gen5L acetyl transferase), chromosome associated factors PCAF complex, STAGA (spt3/TAF/Gen5L acetyl transferase, where its HAT is Gen5L), ADA a transcriptional adaptor, TFIID –contain TAF1, TFTC -TBP free TAF-containing complex, NuA3 and NuA4 (Nucleosomal acetyl transferases of H3 and H4.
It is also interesting to find most of the above said complex does contain some common subunits such as Gcn5, Ada2 and Ada3 which are common to 14 subunit SAGA complex. Likewise Tra1 is common for both SAGA and NuA4,. Tra is a homolog of phosphoinositide 3-kinase (pI3K), it interacts with specific transcriptional activators such as MYC. It is also a fact that several HATs contain one or more TAFs. For example SAGA contains TAF5, 6,9,10 and 12. PCAF contains TAF 9, 10 and 12 but contain their close homologs PAF65b and 65a.

The BRG1 chromatin remodeling protein can associate with numerous chromatin-modifying complexes including transcription coactivators and corepressors. BRG1 (or hBrm) is the central catalytic subunit of SWI/SNF-BAF or -PBAF chromatin remodeling complexes, which have been implicated in the transcriptional activation or repression of a variety of genes. Nuclear receptors can associate with many of these complexes through direct interaction with BAF subunits such as BAF250, BAF60a and BAF57. BRG1 can be found in complexes with transcription coactivators and histone modifying enzymes such as WINAC and NUMAC. Conversely, BRG1 can be assembled in complexes known to repress transcription and induce gene silencing including NCoR and mSin3A/HDAC complexes.
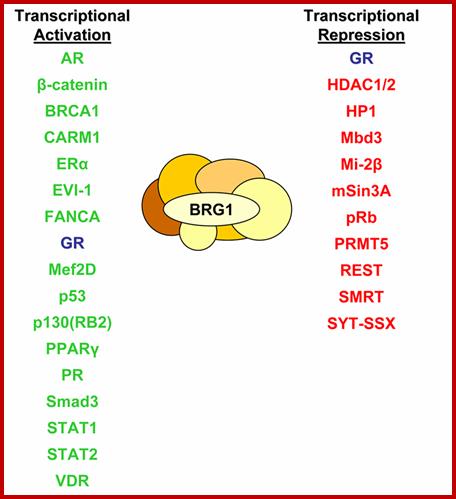
Numerous studies have been conducted using various techniques which have identified BRG1-interacting proteins. For the purpose of this review, these BRG1-associating proteins have been grouped into two categories according to their transcriptional consequence: activation or repression. BRG1 has been reported to associate with numerous proteins implicated in transcriptional activation including various NRs such as AR (Marshall et al., 2003), ERα (Ichinose et al., 1997), GR (Fryer and Archer, 1998), PPARγ (Debril et al., 2004), PR (Vicent et al., 2006) and VDR (Kitagawa et al., 2003). Tumor suppressor proteins have also been found associated with BRG1, including BRCA1 (Bochar et al., 2000), p53 (Lee et al., 2002), and FANCA (Otsuki et al., 2001). Other proteins which are reported to interact with BRG1 include β-catenin (Barker et al., 2001), CARM1 (Xu et al., 2004), EVI-1 (Chi et al., 2003), Mef2D (Ohkawa et al., 2006), p130(RB2) (Giacinti and Giordano, 2006), Smad3 (Xi et al., 2007, 2008) and STAT proteins (Ni and Bremner, 2007; Pattenden et al., 2002). Proteins involved in transcriptional repression also interact with BRG1 and include GR (Bilodeau et al., 2006), HDACs (Underhill et al., 2000), HP-1 (Nielsen et al., 1999), Mbd3 (Datta et al., 2005), Mi-2β (Shimono et al., 2003), mSin3A (Sif et al., 2001), Rb (Giacinti and Giordano, 2006), PRMT5 (Pal et al., 2003), REST (Ooi et al., 2006), SMRT (Jung et al., 2001) and SYT-SSX (Ito et al., 2004; Perani et al., 2003). This list highlights a number of BRG1-interacting proteins which are considered transcription coactivators, corepressors or tumor suppressor proteins
Look at the TAF6, 9 and 12 are the structural homologs of H3, H4 and H2b respectively. Thus one can say they actually form an associated complex to interact with TBP.
Most of the HATs target their components to promoters and they found to be so in active genes. The complex of HATs and their specificity is more intrinsic and it is only now people started to understand the activation of specific gene or group of genes,
The million dollar question what makes specific HATs identify which loci or genes to be acetylated or is it a general phenomenon? The answer lies in the type of HAT and the type of the site identified by certain factors, what are they? Very interesting aspect of HAT associated transcriptional co activators contain (mostly all) contain a ~110 residue module called a bromodomain. Any protein that contains this domain binds to Acetylated Lysine moiety of Histone tails.
For example GCN5 consists of HAT domain followed by a Bromodomain. In the case of TAF1, it has N-terminal kinase domain followed by a HAT domain then two successive tandem Bromodomain. The TAF1 targets specific acetylated histone tails. It contains two nearly identical anti parallel 4 helix bundles, where one finds pockets to recognize acetylated tails and bind to them.
An example to illustrate the activity of a gene shows that the N-terminal tail of H4 contains K residues at 5, 8, 12 and 16; it has been found the acetylation of these said residues increases the transcriptional activity of the said gene,. So the question is what makes these specific sites to be acetylated among the millions of nucleosomes and how the acetylases determine the sites to which bind are promoter elements or other activating sites. Perhaps some specific DNA bound factor provides the site identity for acetylation.
It is also assumed that TAFs1 with its Bromodomain serves to target TFIID to the promoters that are found in the nucleosome containing sequence of a promoter or sequences in the vicinity of a promoter,
The critical feature is that the recruiting HAT-containing co activators complex to a specific upstream element bound by a protein. Once such proteins bind to DNA, the HATs in them can promote acetylation of the nearing nucleosomal Histones and loosen the complex for the assembly of transcriptional complex to initiate transcription or to wait for other activators to act either to activate or suppress the activation.
In all probabilities chances of binding these acetylase complexes to specific sites or regions has to be identified that is already bound to DNA. The logic is simple for all nucleosomes, irrespective the gene promoter they have in them, there should be an identity factor that is bound to DNA in sequence specific manner and such sites have to perpetuate during development. Without such markers, it neighs possible for the HATs to identify the sites.
Histone Deacetylases (HDACs):
In
1996, Dr. S. Schreiber's group was first to clone a mammalian histone
deacetylase (HDAC1). The study revealed that mammalian HDAC1 was related to
yeast transcription regulator RPD3, providing a link between transcription
regulation and histone deacetylation. Several HDACs have since been reported,
including HDAC2 (the mammalian homologue of RPD3) and mammalian HDAC3.
Mammalian HDAC1 and HDAC2, but not HDAC3, are in large multiprotein complexes
containing mSin3, N-CoR or SMRT (corepressors), SAP18, SAP30, RbAp48, and
RbAp46.
Several signal transduction pathways are regulated by the HDAC corepressor
complex. For example, the Sin3A-N-CoR/SMRT-HDAC1, 2complexes is recruited by
unliganded nuclear receptors and the Mad family of bHLH-Zip proteins. An
exciting development is the realization that methyl-CpG- binding protein 2
(MeCP2) binds to Sin3, recruiting more HDAC1/2 complex which could act on
neighboring histones for deacetylation. These reports suggest that histone tail
methylation at arginine and lysine residues and DNA methylation and histone
deacetylation are coupled events in the formation of repressive chromatin
structures and gene silencing.

Composition of HDAC repressor complexes: HDACs lack intrinsic repressor activity and require co-factors for optimal HDAC activity. The co-repressor proteins involved in the major HDAC complexes NuRD (nucleosome remodeling and deacetylase), Sin3 (Switch insensitive 3), Co-REST (Co-repressor of REST (RE1 silencing transcription factor)) and N-CoR and SMRT complexes are shown. NuRD and sin3 complexes share the retinoblastoma associated protein (RbAp)46 and 48 proteins and also contain distinct sets of proteins. Abbreviations: Co-REST, Co-repressor of REST (RE1 silencing transcription factor); MBD3, Methyl CpG binding domain 3; Mi2, Mi2 autoantigen; MTA-2, Metastasis-associated gene family, member 2; N-CoR, Nuclear receptor co-repressor; NuRD, Nucleosome remodelling and deacetylating; RbAp46, Retinoblastoma associated protein of 46 kDa; SAP18, Sin3 associated protein of 18kDa; SDS3, Suppressor of defective silencing 3; Sin3, Switch insensitive 3; SMRT, Silencing mediator for retinoid and thyroid receptors; ZNF217, Zn finger factor 217 kDa.
Adcock et al. Respiratory Research 2006 7:21 doi:10.1186/1465-9921-7-21
It is reported that HDAC1 is associated with MAR- DNA in human breast cancer
cells. These results suggest that HDAC1 may have a role in the organization of
nuclear DNA. It is interesting to note that attachment region binding protein
(ARBP), a nuclear matrix protein that binds to MARs, is homologous to MeCP2.
Thus, N-CoR-Sin3A-HDAC1 complex could be recruited to the nuclear matrix and to
MAR-DNA by MeCP2/ARBP.
Several nuclear factors can
recruit HDAC directly without the assistance of the mSin3, N-CoR and SMRT. HDAC
1, 2 and 3 bind to YY1. Hypo phosphorylated Rb and E2F form a complex with
HDAC1. The recruitment of the E2F-RB-HDAC1 complex is partly responsible for
the repression of the cyclin E promoter in G1 phase of the cell cycle.
Aberrant recruitment of histone modifying enzymes is seen in cancer. For
example, PML-RAR (PLZF-RAR) and AML-1-ETO, oncoproteins in acute promyelocytic
or myeloid leukemia generated by chromosomal translocations, recruit
SMRT-mSin3A-HDAC1 and N-CoR-mSin3A-HDAC1, 2 complexes, SMRT-mSin3A-HDAC1
complex is recruited by the BTB/POZ domain found in the oncoproteins LAZ3/BCL6.
Isolation of novel mammalian PRMTs by molecular cloning and novel substrates (histones and non-histone proteins) and determining the corresponding methylation sites to define novel gene regulatory pathways in which this posttranslational modification is involved is important. With respect to the histone code hypothesis it is now known whether site-specific arginine methylation interferes/cross-talks with other post-translational modifications and how this modification is "translated" into chromatin alteration on the molecular level. Discovery of reverse arginine methylation by specific demethylase therefore it is a dynamic process as it has been shown to be the case for other histone modifications.
Histone Deacetylases by removing acetyl group allow other components to bind or otherwise makes the region inaccessible for the assembly of transcriptional components including PIC or BTA and other upstream factors that regulated the gene expression; thus repress the expression a gene. Deacetylation of specific lysine otherwise of histone tail, provides an opportunity for other enzymes to join and modify the tails in different and generate different codes. Example, methylases that methylate histone tail at specific amino acid residues, which need not be the same residue that is deacetylated. They can be different. Among the many; ten HDAs have been identified with certainty. There are ten HDAs in yeast, 17 in humans. HDAs have a family of proteins such as Class I and Class 2 etc. There are at least three classes- such as Class I, Class II and Class III; most of them are multisubunit complexes.
In humans the HDACs consist of the said three classes: Class I-contain HDAC 1, 2, 3 and 8. Class II consists of HDACs 4, to 7 and 10. And the Class III contains Sirutins (SIRT1-7) (SIR for Silent information regulators). Most of them are multisubunit complexes, where some are common to all the classes and some are specific to each of the classes. Some of the components are –Sin 3, NurD (nucleosome remodeling histone deacetylases), Co rest (co repressor of RE1 silencing transcription factor), NCOR (nuclear hormone receptor co repressor), SMRT (silencing mediator of retinoid and thyroid hormone receptor), Most of them serve as transcriptional co repressors at different sites and with different composition. For example REST on binding to its target, recruits CoRest and SIN3, which together repress the expression of the gene where they bind. Such methylated complexes produce compaction of the chromosomal loci and make it heterochromatic, which can be discerned Giemsa staining.
Histone Methylases and Demethylase:
Lysine and Arginine residues in H3 and H4 tails are the targets for methylation by Histone methyl transferases (HMTs). They use S Adenosyl Methionine, SAM as the methyl donor. The enzymes have SET domain [(Su (var) 3-9, E (Z), Trithorax], contain catalytic sites. Interestingly no demethylase have been identified, which means methylation is not reversible (?). But in some cases trimethylation of a histone tail is demethylated to di methylated sites. Recent investigations have shown that demethylase exist. Methyl groups are removed by specific demethylases.
Methylated histone tails are recognized by proteins containing Chromodomain. For example methylated H3 at lysine 9 is recognized by Chromodomain containing Heterochromatin 1 (HP1) protein. The binding of HP1 recruits other proteins to control chromatin structure and gene expression. The said enzymes have active sites as clefts or grooves where only such tails bind with specific methylated amino acid (s). This binding can lead to spreading to nearby chromatin to be silenced. This happens due to to other domain called Chromodomain shadow domain of HP1 is associated with methyl transferase (recruits HMT Suv 39h protein complex) that methylates the neighboring histone tails, which recruits more HP1 proteins.
Thus heterochromatin spreads. However the spreading heterochromatization is often checked by insulators found in the pathway of heterochromatization. ex – chick b-globin clusters recruit HATs that acetylates H3 lys9 at nearby nucleosomes, thus it checks the spreading of heterochromatization. Question is what makes the methylation mediated HP1 binding leads to heterochromatization or what we call it as condensation of chromatin?
Histone Ubiquitination and Transcription:
Ubiquitination to protein via lysine ligation leads to proteasome mediated protein degradation. But in yeast ubiquitination of H2B Lys 123 mediated by ubiquitin ligase i.e. Rad6 and E3 Bre1 are responsible for methylation of H3 K4 and k79. This modification results in silencing genes located near telomere. It is suggested that H2B ubiquitination (not for destruction) functions as master switch that controls selective methylation and silencing telomeric gene silencing. It is interesting to find that TAF1 subunit of TF IID is involved in post translational modification of has been found in Drosophila. It appears that ubiquitination of certain Histones sites is emerging as regulators.
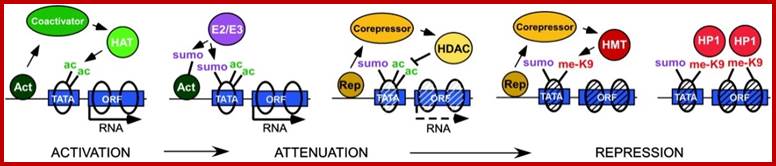
Model for sumoylation function in transcription: Horizontal line represents a gene with a TATA box-containing promoter and ORF; ovals represent histone octamers/nucleosomes. Through a co activator, a DNA-bound activator can recruit a histone acetyltransferase (HAT) that acetylates histones and promotes chromatin structure amenable to transcription. This acetylation can potentially recruit SUMO-conjugating enzymes (E2/E3) capable of modifying either histones or activators to give an attenuating effect. A corepressor and HDAC activity could then be recruited by a DNA-bound repressor (possibly even with SUMO contributing to the interaction), deacetylation of histones, and making way for the addition of repression-specific methylation marks, such as H3 K9-methyl, by an HMT. Finally, methylated histones (and possibly SUMO) would recruit HP1, contributing to chromatin structure in a static repressed state.

The SUMO modification of proteins in general is required for normal chromosome condensation and mitosis. Disruption of sumoylation pathway leads to mitotic defects and embryonic development failure characterized by inability of cells to properly condense chromatin. We pursue a hypothesis that mitotic chromatin condensation and heterochromatic gene silencing are intrinsically linked at the biochemical level, both being dependent on SUMO modification of structural proteins and enzymatic machinery shared by these processes.
Possible Roles of modified Histones (from Harper):
1. Acetylation of
histones H3 and H4 is associated with the activation or inactivation of gene
transcription. 2. Acetylation of core histones is associated with chromosomal
assembly during DNA replication.3. Phosphorylation of histone H1 is associated
with the condensation of chromosomes during the replication cycle.
4. ADP-ribosylation of histones is associated with DNA repair.
5. Methylation of histones is correlated with of activation and repression gene
transcription. 6. Mono-ubiquitylation is associated with gene activation,
repression, and heterochromatic; gene silencing. 7. Sumoylation of histones
(SUMO; small ubiquitin-related modifier) leads to transcription repression.
Chromatin Remodelling and Regulation of Gene Expression:
In prokaryotes bacterial circular DNA is also compacted by histone like proteins; but DNA in eukaryotes is compacted into chromatin; this compaction is essential for the DNA is very long as 3.2x10^9bp in humans or more in some 10^12bp. Such a long and so thin 10.5A thick thread cannot be subjected to such changes during cell division, repair and recombination. The DNA has to go through opening and closing at different sites may be in thousands during transcription. In order to protect the DNA from shear and wear it is stabilized and strengthened by histones, which compacts DNA to 30nm thread and with the association of scaffold proteins it is further strengthened and compacted to 300nm fibre, which remains so through the cellular changes except DNA replication. Rest of the time this 300nm fibre very often undergoes regional compaction at different loci or regions. Such compactions are called heterochromatin and uncompacted 300n fibre is called euchromatin, which is engaged in transcription. The 300nm fibre is bound by thousands of nonhistone proteins; that makes chromosome visible structure. This chromatin exists in relaxed state at interphase where its HC domains remain bound to inner nuclear membrane matrix proteins. Some region of interphase chromatin are relaxed and involved in transcription, but there are regions where the 300nm chromatin is further condensed by heterochromatization, which can be constitutive (pericentric and telocentric regions) or facultative (its position varies in different tissue types. The heterochromatic condensation is greater than metaphase condensation. Amount of DNA per unit in HC is more than metaphase compacted chromatin. The HC regions can be visualized by differential staining (Giemsa) of metaphase chromosomes.
DNA methylation: Fingerprints of (epi) Genome:
DNA methylation takes place in specific DNA sequences such as 5’-CpGpCpGpCpG-3’. Such 5-CpG-3’ repeats can be found 500 base pairs long and are located mostly within gene 5’promoters regions of genes. Such repeat sequences are called CpG islands. Such groups called islands can be used as finger prints of a genome. Methylation of Cytosine at 5’ position takes place by Cytosine methyl transferase using S-Adenocylmethionine (SAM) as the donor. Such CpG islands are also found in other regions such as coding regions and non-coding regions too. Such CpG repeat islands are located in different parts of the genome and it is a fixed pattern of the genome. But such repeats in gene promoter region get methylated 5’C-CH3-G3’.

Developmental Biology; We
are studying common mechanisms of chromatin remodeling, DNA methylation and
transcriptional regulation in lung development that might be activated in lung
diseases, Directed by Wellington V. Cardoso, MD, PhD;http://www.bumc.bu.edu/
DNA methylation is performed a group of methylases called DNMTs. There are DNMT1, DNMT3a and DNMT3b. The DNMT1 performs methylation of hemimethylated DNA strand during replication. There are enzymes which remove such methyl groups from CpG islands. Such DNMTs inhibitor is Azacytidine. Treatment with Azacytidine makes the genes which are inactive become active. Methylation of 5-CpG-3 leads to the recruitments of specific proteins such as MeCP2, which recruits HDAC. Such methylation of 5’-CpG-3 islands in promoter regions renders the gene inactive. This is achieved by recruiting HDACs and heterochromatin binding proteins such as HP1 etc. This makes the gene promoter inaccessible for transcriptional factors and enzymes.
In most cases, methylation of DNA is a fairly long-term, stable conversion, but in some cases, such as in germ cells, when silencing of imprinted genes must be reversed, demethylation can take place to allow for "epigenetic reprogramming." The exact mechanisms for demethylation are not entirely understood; however, it appears that this process may be mediated by the removal of amino groups by DNA deaminases (Morgan et al., 2004). After deamination, the DNA has a mismatch and is repaired, causing it to become demethylated. In fact, studies using inhibitors of one DNMT enzyme showed that this enzyme was involved in not only DNA methylation, but also in the removal of amino groups; http://www.nature.com/scitable/topicpage.

Methylation modification of DNA at the 5-carbon position of cytosine by DNMTs where SAM donates the –CH3 group and is converted to SAH. This reaction is potentially reversible by a yet to be defined DNA methylase.
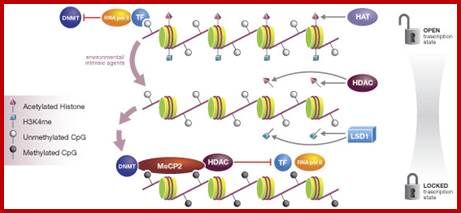
A permissive state for transcription includes histones acetylated by HAT as well as methylated at H3K4 (H3K4me). Unmethylated CpGs are bound by transcription factors and RNA polymerase II thereby blocking interaction with DNMT. Environmental influences potentially trigger reversal of acetylation by HDAC and removal of methylation by LSD1. In this state, CpGs are vulnerable to methylation by DNMT and are bound by MeCP2, which recruits HDAC. HDAC maintains a deacetylated state of the histone, locking the chromatin in a repressed state that prohibits transcription factor binding. Presently the precise order of these events is unclear.

Chromatin structure regulates transcriptional activity: Histone-deacetylase inhibitors: novel drugs for the treatment of cancer; Nucleosomes consist of DNA (black line) wrapped around histone octomers (purple). Post-translational modification of histone tails by methylation (Me), phosphorylation (P) or acetylation (Ac) can alter the higher-order nucleosome structure. Nucleosome structure can be regulated by ATP-dependent chromatin remodellers (yellow cylinders), and the opposing actions of histone acetyltransferases (HATs) and histone deacetylases (HDACs). Methyl-binding proteins, such as the methyl-CpG-binding protein (MECP2), target methylated DNA (yellow) and recruit HDACs. a | DNA methylation and histone deacetylation induce a closed-chromatin configuration and transcriptional repression. b | Histone acetylation and demethylation of DNA relaxes chromatin, and allows transcriptional activation, Ricky W. Johnstone; http://www.nature.com/

DNA Methylation: The diagram shows a representative region of
genomic DNA in a normal cell. The region shown contains repeat-rich,
hypermethylated pericentromeric heterochromatin and an actively transcribed
tumour suppressor gene (TSG) associated with a hypomethylated CpG island
(indicated in red). In tumour cells, repeat-rich heterochromatin becomes
hypomethylated and this contributes to genomic instability, a hallmark of
tumour cells, through increased mitotic recombination events. De novo methylation of CpG islands also occurs
in cancer cells, and can result in the transcriptional silencing of
growth-regulatory genes. These changes in methylation are early events in early
tumorogenesis © 2005 Nature Publishing Group
Robertson,K. DNA methylation and human disease . Nature Reviews Genetics 6, 598’ ![]()
There are many ways that gene expression is controlled in eukaryotes, but methylation of DNA (not to be confused with histone methylation) is a common epigenetic signaling tool that cells use to lock genes in the "off" position. In recent decades, researchers have learned a great deal about DNA methylation, including how it occurs and where it occurs, and they have also discovered that methylation is an important component in numerous cellular processes, including embryonic development, genomic imprinting, X-chromosome inactivation, and preservation of chromosome stability. Given the many processes in which methylation plays a part, it is perhaps not surprising that researchers have also linked errors in methylation to a variety of devastating consequences, including several human diseases. Experiments with 5-azacytidine provide early clues to the role of Methylation in gene expression. DNA methylation occurs at the cytosine bases of eukaryotic DNA, which are converted to 5-methylcytosine by DNA methyltransferase (DNMT) enzymes. The altered cytosine residues are usually immediately adjacent to a guanine nucleotide, resulting in two methylated cytosine residues sitting diagonally to each other on opposing DNA strands. Methylation can be observed by staining cells with an immuno fluorescent labeled antibody for 5-methylcytosine. In mammals, methylation is found sparsely but globally, distributed in definite CpG sequences throughout the entire genome, with the exception of CpG islands, or certain stretches (approximately 1 kilobase in length) where high CpG contents are found. Note the methylation of these sequences can lead to inappropriate gene silencing, such as the silencing of tumor suppressor genes in cancer cells. results of immunoprecipitation studies using human cells suggest that DNA methylation and histone methylation work together during replication to ensure that specific methylation patterns are passed on to progeny cells (Sarraf & Stancheva, 2004).
In an interestingly coordinated process, proteins that bind to methylated DNA also form complexes with the proteins involved in deacetylation of histones. Therefore, when DNA is methylated, nearby histones are deacetylated, resulting in compounded inhibitory effects on transcription. Likewise, demethylated DNA does not attract deacetylating enzymes to the histones, allowing them to remain acetylated and more mobile, thus promoting transcription. In most cases, methylation of DNA is a fairly long-term, stable conversion, but in some cases, such as in germ cells, when silencing of imprinted genesmust be reversed, demethylation can take place to allow for "epigenetic reprogramming." The exact mechanisms for demethylation are not entirely understood; however, it appears that this process may be mediated by the removal of amino groups by DNA deaminases (Morgan et al., 2004). After deamination, the DNA has a mismatch and is repaired, causing it to become demethylated. In fact, studies using inhibitors of one DNMT enzyme showed that this enzyme was involved in not only DNA methylation, but also in the removal of amino groups. Theresa Phillips, Ph.D. (Write Science Right) © 2008 Nature Education http://www.nature.com/
Silent Chromatin in the Middle – “Centromere”:
Central Centromere is bordered by a distinct heterochromatin a dominant region of the chromosome microscopically visible. It looks like a not stainable gap as if there is no DNA in that region. Centromere plays a par excellent role in chromosomal movement during mitosis and meiosis. This acts as core structural component for the binding of Kinetochore complex and for the tractile fibers to attach and pull sister chromatids or homologous chromosomes to their respective poles. The central region of centromere is bound by histone octamers but one of the histone is different called CEP-A/B. This part of the nucleosomes is free from heterochromatization; how this region is left free from heterochromatization while the borders are distinctly heterochromatic very condensed structure? This is designed for the binding of kinetochore during metaphase. Is the early kinetochore element prevents such heterochromatization? On either side of central centromere one finds a distinct heterochromatin. Its HC formation is similar to telomere where H3K9 deacetylation and its methylation lead to the binding of HP1; this spreads to neighbor nucleosomes.

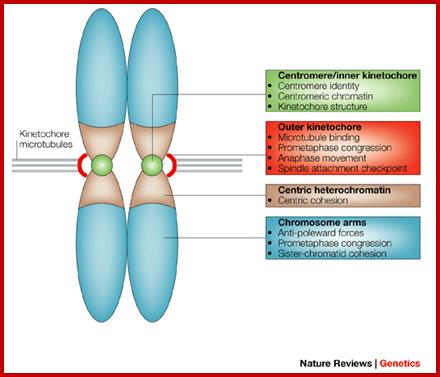
Structurally the CEN region appears as a gap (non-stainable), but on either side of the CEN region one finds darkly stainable chromatin called constitutive heterochromatin; none of the genes in the region are active but silent. Structurally the CEN region is made up of satellite DNA for that matter most of constitutive heterochromatin is made up of satellite DNA (?). Centromeric (CEN) chromatin is embedded in heterochromatin and contains blocks of histone H3 nucleosomes interspersed with blocks of CENP-A nucleosomes, the histone H3 variant that provides a structural and functional foundation for the kinetochore. The spectrum of histone modifications present in human and Drosophila melanogaster CEN chromatin is distinct from that of both euchromatin and flanking heterochromatin.

A model for DNA bundling by the CENP-B dimer in centromeric chromatin: Orange ribbons with arrowheads indicate 171-base pair α-satellite repeats. Green circles show the centromeric nucleosomes, which may contain histones (H2A, H2B, and H4) and CENP-A. The crystal structures of the dimerization and DNA-binding domains of CENP-B are connected by the Pro-rich region (yellow dashed lines), the central transposase-like domains (pink dashed circles), and the Asp/Glu-rich region-blue dashed lines
This is the CENP-B that is associated with CEN DNA
;)
The kinetochore can be thought of as three sets of subcomponents. a) The chromosomal DNA-inner kinetochore protein interface. b) The inner kinetochore-mitotic spindle interface. c) The kinetochore protein-cell cycle machinery interface. APC anaphase-promoting complex; CEN, centromeric DNA; SCF, ubiquitin-ligase complex. Copyright 2007 Nature Publishing Group, Kitagawa, K., et. al., The spindle-assembly checkpoint in space and time, Nature Reviews Molecular Cell Biology 2, 678-687.
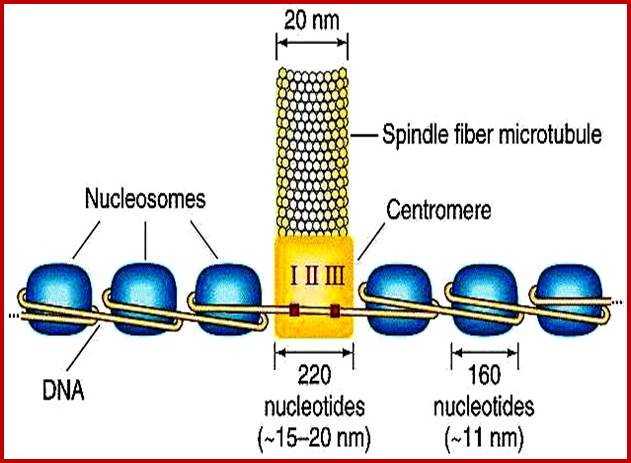
http://www.cbs.dtu.dk/
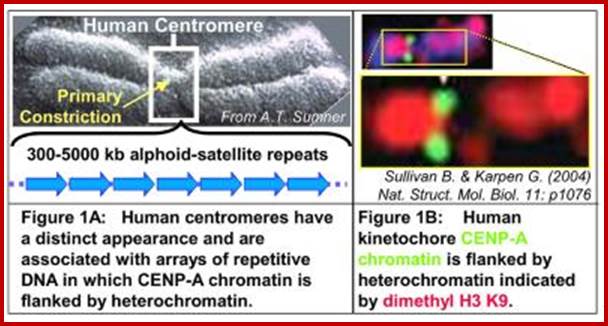
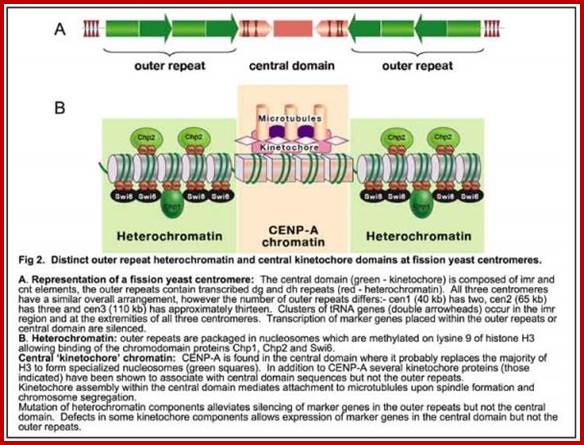

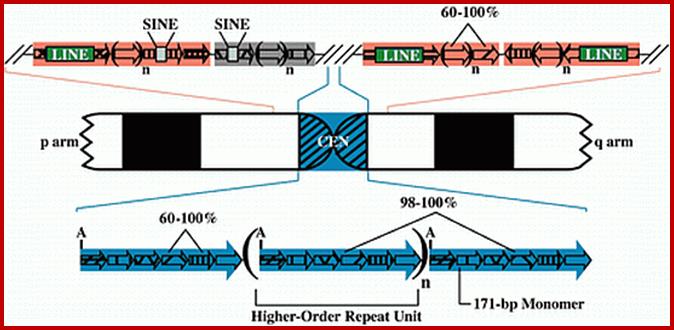
Centromere DNA consists of tandem repeats of LINES and SINES and AT rich DNA.
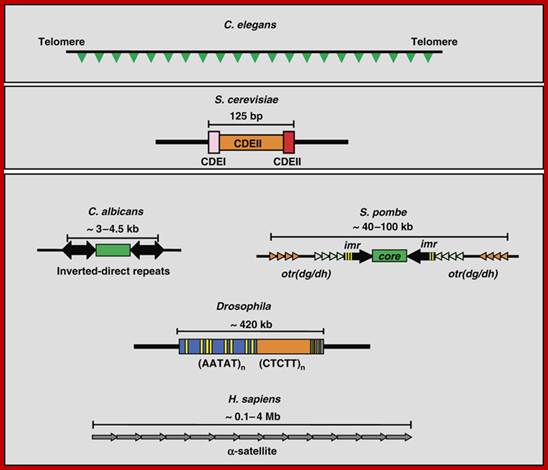
Tandem repeat sequence of DNA at telomeres and CEN regions

Figure: Proposed structure for centromere DNA at the kinetochore region. (Top) Bi-oriented sister chromatids adopt a cruciform structure. Centromere-flanking chromatin is held together by intrastrand cohesin bridges and chromosome arms by interstrand cohesin bridges. The transition between these two regions in budding yeast is mobile and on average 7 kb from the centromere core. (Bottom) The Cse4-containing nucleosome (orange circle) and flanking nucleosomes (green circles) are proximal to the microtubule plus-end.
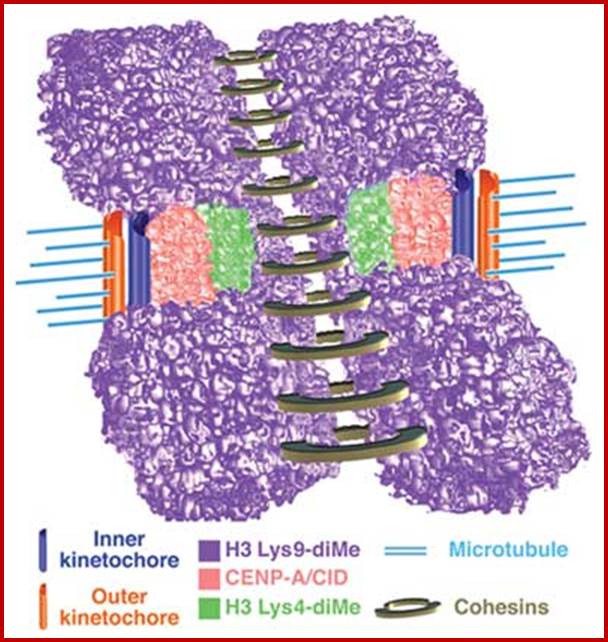
Figure: Model for three-dimensional organization of centromeric (CEN) chromatin in D. melanogaster and humans.
Incorporation of the two-dimensional and three-dimensional histone modification
patterns extends our understanding of the chromatin composition and
organization of the CEN region, and suggests that interspersed CENP-A/CID and
H3 Lys4-dimethyl nucleosome blocks comprise a unique chromatin state that is
distinct from the flanking heterochromatin. Associations between similarly
modified nucleosome blocks are proposed to contribute to the formation of
distinct three-dimensional structures in CEN and flanking chromatin.
Interspersed CENP-A/CID and distinctly modified H3 and H4 may mediate formation
of the 'cylindrical' three-dimensional structures observed in metaphase
chromosomes. H3 Lys9-diMe chromatin, which recruits heterochromatin proteins
such as HP1 and cohesion proteins such as RAD21/SCC1, is present in the inner
kinetochore space between mitotic sister chromatids and in regions that flank
CEN chromatin. This arrangement may position CENP-A toward the pole-ward face
of the mitotic chromosome and facilitate recruitment of outer kinetochore
proteins, and promote HP1 self-interaction and proper chromosome condensation
and cohesion. Cohesins are presented as ringed structures, in accord with
recent models.

Fig: Model for the kinetochore. CenH3 hemisomes (red/gray disks) are separated by extended linker DNAs and so are decondensed relative to surrounding heterochromatin (blue disks). Asymmetric CenH3 nucleosomes assemble in random orientations [CenH3/H4 (red) and H2A/H2B (gray)]. Only one unit of a CenH3-rich block is shown. During mitotic condensation, heterochromatin packs tightly as a result of its homogeneity. Intervening blocks of CenH3 chromatin cannot pack into this crystal-like structure because of its smaller size, long linkers, and heterogeneity in its relative orientation, resulting in extruded loops of uncondensed CenH3 nucleosomes that serve as the foundation for kinetochore formation. The flanking gray cones represent pericentric regions flanking the primary constriction.

Schematic depicting centromeric chromatin composition in relation to the cell cycle. CENP-A–containing nucleosomes (red) are interspersed with canonical H3-containing nucleosomes (green) after replication in S phase, and this mixed set of nucleosomes is the substrate for nucleating kinetochore assembly in mitosis and is maintained as cells exit in anaphase. CENP-A assembly initiates in telophase and proceeds through early G1 (presumably concurrent with removal of H3 nucleosomes). CENP-A– and H3-containing nucleosomes are stylized as single nucleosomes but may represent continuous alternating arrays of one or the other type. In mitosis, CENP-A nucleosomes may coalesce to form a rigid interface for kinetochore formation as proposed previously (Zinkowski et al., 1991; Blower et al., 2002; Black et al., 2004,2007a,b
Telomeric Region and its Repression:
Telomeric silencing requires proteins such as RAP1 that binds to DNA and Sir (Silent information Regulator) proteins bind to RAP (Ras related Protein); RAP acts as nucleation centre. Sir2 is deacetylase protein. Proteins such as Sir1, 3 and 4 associate as a complex in telomeric region and silence the genes, if any. And HMR (yeast mating Region?) region in yeast also performs in similar fashion. Telomeric tail lacks histones. In the proximal region Histone H3 and H4 tails Sir3 and Sir4 bind to histone tails and maintain the tails in deacetylated state. Hypo-acetylated regions are associated with Sir2; such complexes bound to each other and prevent the entry of any transcriptional regulator proteins.
Repression of certain gene loci such as HML and HMR is due to condensation of chromatin in promoter-regulator regions. Adenine methylases gene introduced into yeast cell methylates GATC sequences in MAT region but not HML and HMR for they are already compacted for the enzyme to act.
Chromosomes with telomeric ends
Any gene placed near telomere gets silenced. RAP1 and SIR proteins bind to telomere region for repression. RAP1 binds to Telomere single stranded DNA (extended) sequences, which are tandemly repeated. SIR2 is HDAC, RAP1 binds to DNA, SIR3 and SIR4 bind to RAP1 and Sir2 binds to Sir4 ; These RAP1 and Sir proteins bind to hypo acetylated histones; this association spread to neighbor histone bound telomeric regions. Sir3 and 4 bind to hypoacetylated H3 and H4 tail and Sir2 maintains them in deacetylated state. Fluorescence labeled probes show that telomeric regions are condensed state like other Heterochromatin bands. A similar programme is operated in pericentric region; however there is predominance of H3H4 modification operates in CEN region.
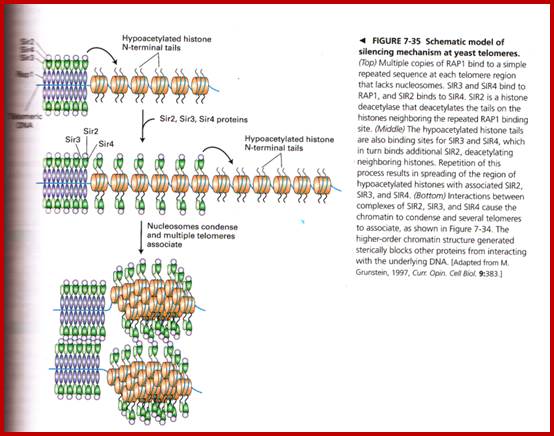
Chromatin- compacted and silent
Silent chromatin at the ends: lessons from yeasts: Marc Bühler and Susan M Gasser.

Heterochromatin in S. cerevisiae, S. pombe, P. falciparum and human

Figure Hypothetical model for heterochromatin assembly at P. falciparum chromosome ends. This is a general view of the known chromatin components at P. falciparum sub telomeres. The spreading of heterochromatin along the different TAREs into adjacent coding regions probably involves PfHP1, PfSir2 and PfKMT1 in cooperation. The role of PfOrc1 in this process remains unknown.
In most organisms, histones H3 and H4 are among the most conserved proteins. Their amino terminal NH2 terminal tails, which are necessary for the formation of the nucleosomal core, are fairly conserved in terms of their sequence, particularly at residues that are susceptible to specific covalent modifications. The amino terminal ends of histones H3 and H4 of P. falciparum contain sites susceptible to post-translational changes. Several modifications have been identified in P. falciparum by mass spectrometry analysis of histones, Western blot assays performed with antibodies specific to methylated and acetylated histones, and ChIP on ChIP assays. Those modifications are: acetylation of histone H3 at residues K9, K14, K18, and K27 and of histone H4 at K5, K8, K12 and K16; methylation of histone H3 at K4, K9 and K36 and histone H4 at K20, as well as the sumoylation of histone H4. Recently, a comprehensive mass spectrometry analysis of P. falciparum histones identified 44 new post-transcriptional modification sites in these proteins, most of them associated with a transcriptionally permissive state.
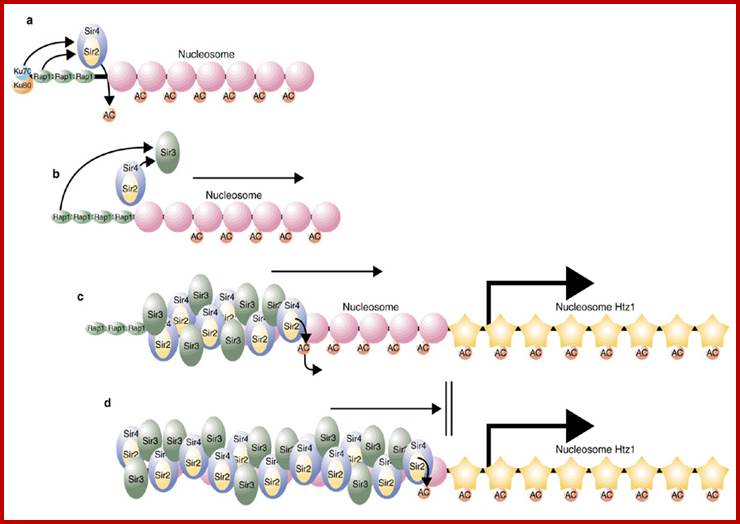
a, Telomeric heterochromatin is nucleated by the interaction of Sir2–Sir4 with telomere-binding proteins. b, After the deacetylation of histone tails by Sir2, Sir3 is recruited. c, Propagation of heterochromatin caused by further rounds of histone deacetylation and binding of Sir protein. d, HTZ1-containing nucleosomes represent a barrier and protect active regions from being silenced.
Schematic model of silencing mechanism at yeast telomeres:
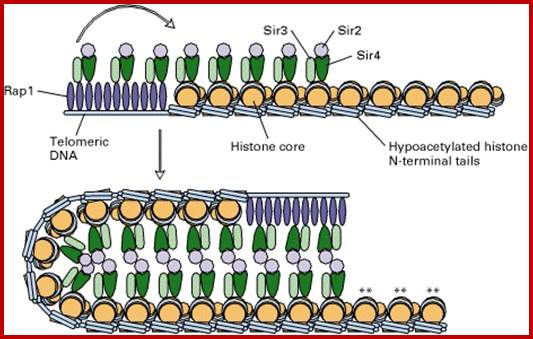
Telomere consists of repeat sequence of TTAGGG with single stranded 3’ tail. The length of this sequence shrinks and expands in time. This sequence is protected from exonuclease activity by heterochromatization. The 3’ single stranded tail is bound by RAP1 protein which gets associated with Sir proteins. Posterior to this tail one finds dsDNA associated nucleosomal structure, which is also bound by Sir3 and Sir4 to hypoacetylated and methylated histone tail and Sir2 a deacetylase is associated with Sir4; interaction between Sir proteins leads to looping and heterochromatization (compaction).
Silencing at the telomeres
Telomeres can be found at the ends of all 16 chromosomes in yeast for that matter in all chromosomes and in all organisms. They consist of a 300-350 bp region with irregular TG1-3 repeats, which are disrupted by X- and Y-elements of variable size (Palladino and Gasser, 1994). The Rap1 binds on average every 18 bp in the telomeric repeats and is a structural core component of the telomeres. The Rap1 binding region is also referred to as the telosome and is characterized by its nuclease resistance.
Rap1 in turn recruits Sir2, Sir3 and Sir4 to the telomeres, where Sir3 and Sir4 bind to the N-termini of histone H3 and H4. The Sir proteins spread in a histone-dependent manner up to 2.8 kb into the core telomeric heterochromatin. (Strahl-Bolsinger, et al., 1997) have proposed a model in which telosomal Rap1 folds back onto subtelomeric regions. This allows a condensation at telomeric heterochromatin due to the interaction between Rap1 and the Sir’s, as well as among the Sir proteins themselves.
Telomeres: protecting chromosomes against genome instability
Roderick J. O'Sullivan & Jan Karl Seder
G1 phase telomeric chromatin has hallmarks of constitutive heterochromatin: the trimethylation of Lys9 of H3 (H3K9me3) and of Lys20 of H4 (H4K20me3) and heterochromatin protein 1 (HP1) binding (see the figure, part a, right). Subtelomeric chromatin can be distinguished by a regular nucleosomal distribution, extensive DNA methylation and histone post-translational modifications that are distinct from those at telomeres (part a, left). The replication of DNA during S phase coincides with the disruption and restoration of the parental chromatin identity. Newly synthesized histones are acetylated at Lys5, Lys12 and Lys16 of H4 and at Lys9 and Lys56 of H3. The removal of these acetyl groups at telomeres by SIRT proteins seems to be important for regulating the association of accessory proteins to telomeres, as exhibited by the relationship between the acetylation of Lys9 of H3 (H3K9ac), SIRT6 and Werner syndrome ATP-dependent helicase (WRN) function.
Changes in chromatin structure have been shown to occur at dysfunctional telomeres. These changes are often similar to those exhibited at sites of DNA damage, such as phosphorylation of H2AX, changes in H4K20me2 levels and recruitment of tumour suppressor p53-binding protein 1 (TP53BP1), implying that there is a general 'epigenetic' stress response (see the figure, part c). However, it is still unclear whether changes seen at dysfunctional telomeres are proactive or merely responsive to changes in telomeric architecture. Nuclear reprogramming also leads to dramatic changes in telomeric chromatin and telomere length, emphasizing the dynamic and developmentally regulated nature of chromosome ends. POT1= protection of telomeres 1; TERRA= telomeric repeat-containing RNA; TRF2= telomeric repeat-binding factor 2.
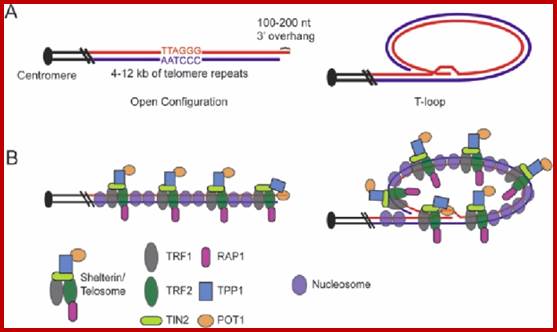

Telomeric organization
Heterochromatins:
|
Constitutive HC |
Facultative HC
|
|
Rich in satellite DNA, polymorphic |
Not enriched in satellite DNA-non polymorphic |
|
HC is stable and conserves its heterochromatic properties during all stages of development and in all tissues |
Facultative HC is reversible, that is to say, it can change from the heterochromatic state to a euchromatic state in different issues |
|
HC is highly polymorphic, no phenotypic effect |
The facultative HC is not particularly rich in satellite DNA, and is therefore not polymorphic. |
|
HC is strongly stained by the C-band technique. This staining could be the result of the very rapid renaturation of the satellite DNA following denaturation. |
HC is not stained by the C-band technique. |
|
Constitutive heterochromatin deeply stained by DAPI |
Less stained |
|
Exhibits C-bands |
No C-bands |
|
|
|
|
Heterochromatin (HCh) |
Euchromatin (EuCh) |
|
HCh is condensed, and nucleosomes are compacted
|
EuCh relaxed
|
|
Heterochromatin DNA is late in replicating,
|
Replicates early |
|
Heterochromatin DNA is methylated,
|
EuCh histone3 methylated at H3K4 (in active regions) |
|
In heterochromatin, histones are hypo-acetylated:
|
EuCh is hyper acetylated |
|
Histones from heterochromatin are methylated on H3 lysine 9,
|
Histone 3 methylated at K4 |
|
Heterochromatin is transcriptionally inactive,
|
Transcriptionally active |
|
Heterochromatin does not participate in genetic recombination,
|
Involved in genetic recombination |
|
Heterochromatin has a gregarious instinct,
|
Normal |
|
Certain RNA such as Xist RNA and few others are responsible heterochromatization |
Some RNAi are involved in facultative HCh |
|
HCh is involved epigenetic and genetic imprinting phenomenon |
EuCh not involved in epigenetic, but facultative Hc can |
|
Defective HCh in sex vesicles cause hypofertility or a sterility |
HCh causes diseases such as ICF and Roberts syndrome etc |
|
|
|
Heterochromatin in General:

The basic unit of chromatin organization is the nucleosome, which comprises 147 bp of DNA wrapped around a core of histone proteins. Nucleosomes can be organized into higher order structures and the level of packaging can have profound consequences on all DNA-mediated processes including gene regulation. Euchromatin is associated with an open chromatin conformation and this structure is permissible for transcription whereas heterochromatin is more compact and refractory to factors that need to gain access to the DNA template. Nucleosome positioning and chromatin compaction can be influenced by a wide-range of processes including modification to both histones and DNA.
The nucleosome, made up of four histone proteins (H2A, H2B, H3, and H4); they are the primary building blocks of chromatin. Originally it was thought to function as a static scaffold for DNA packaging. Histones, more recently been, have shown to be dynamic proteins; undergoing multiple types of post-translational modifications. Two such modifications, methylation of arginine and lysine residues are major determinants for formation of active and inactive regions of the genome. Arginine methylation of histones H3 (Arg2, 17, 26) and H4 (Arg3) promotes transcriptional activation and is mediated by a family of protein arginine methyltransferases (PRMTs), including the co-activators PRMT1 and CARM1 (PRMT4). In contrast, a more diverse set of histone lysine methyltransferases has been identified, all but one of which contain a conserved catalytic SET domain originally identified in the Drosophila Su[var]3-9, Enhancer of Zeste, and Trithorax proteins. Lysine methylation has been implicated in both transcriptional activation (H3 Lys4, 36, 79) and silencing (H3 Lys9, 27, H4 Lys20). Histone H3 and H4 acetylation is common in active chromatin. These modifications for gene activation or repression are mostly found in the upstream of the promoter and promoter regions.

Euchromatin in certain regions become heterochromatin and vice versa; histone deacetylation, histone methylation and the binding of corepressor like HP1 leads to heterochromatization; on the contrary demethylation and acetylation, phosphorylation, and specific H3K4 methylation and loss of H1 lead to euchromatization.
The figure shows activation and inactivation by specific histone modification and association of repressor and activator complexes. Histone modification show changes in the compaction and relaxation of nucleosomal thread. Binding of activator or repressor to certain sequences act as nucleating centers for gene activation or repression respectively.

Schematic depiction of the role of histone acetylation in chromatin remodeling: Acetylation of histone carboxyl-terminal tails is believed to promote unpacking of nucleosomes from 30-nm chromatin fibers. Histone PTM enzymes or agents that can influence the state of chromatin packing are indicated as favoring either side of this reversible process. Possible roles of phosphorylation and ubiquitination are less well understood because these effects may be dependent upon modifications of specific amino acids or influenced in the context of other modifications concurrently existent histones.

Oncogeneic fusion proteins such as AML1–ETO, CBFB–MYH11 and PML–RARA recruit transcriptional co-repressor complexes (including nuclear receptor co-repressor 1 (NCOR1) and NCOR2) that result in the loss of histone acetylation and the acquisition of repressive histone modification marks, such as histone H3 lysine 9 (H3K9) methylation and H3K27 trimethylation, as well as DNA methylation, and thereby a closed chromatin structure. This leads to the transcriptional silencing of various target genes, including genes that are crucial for haematopoietic differentiation. Epigenetic or transcriptional therapy (targeting the fusion proteins, components of the co-repressor complexes and downstream effectors such as microRNAs) has the potential to reverse these changes, leading to histone acetylation, acquisition of active marks such as H3K4 methylation, an open chromatin structure with subsequent transcriptional activation and differentiation of the leukemic clone. Ac, histone acetylation; AML1, acute myeloid leukemia 1; CBFB, core binding factor-β; CpG, cytosine residues that precede guanosine; DNMT, DNA methyltransferase; HDAC, histone deacetylase; HMT, histone methyltransferase; PML, promyelocytic leukemia; RARA, retinoic acid receptor-α; RNApol II, RNA polymerase II; TF, transcription factor

One of the main experimental techniques that has allowed the elucidation of chromatin structure and function is chromatin immunoprecipitation (ChIP). The first step in ChIP analysis is to cross-link the chromatin associated protein to DNA in live cells. The cells are then lysed and DNA complexes are sheared into small fragments, and the protein of interest is immunoprecipitated. Consequently, DNA sequences that interact with the protein of interest are enriched in this immunoprecipitation stage. Protein–DNA cross-links are subsequently reversed and the amount of DNA precipitated is quantified, usually by real-time PCR or microarray analysis. Thus, ChIP analysis, using antibodies to specific chromatin-associated proteins, together with an analysis of individual histone modifications, enables the characterization of endogenous chromatin structure at individual genomic regions.
Methylation and Acetylation of Histone in Specific Regions of the Nucleosomes is the Key for Gene Expression:
Localized gene silencing:

In eukaryotes, DNA is wrapped around histones to form nucleosome, a fundamental unit of chromatin. The native chromatin is further compacted into higher-order structure and plays a critical role in various chromosomal processes, such as gene regulation, recombination, replication, chromosome condensation and the proper segregation of chromosomes. The organization of higher-order chromatin structure has been linked to the post-translational modifications of histones, such as acetylation, phosphorylation and ubiquitination. How these histone modifications participate in the modulation of chromatin structure has remained elusive. In their recent publication in Science, HFSP Long-term Fellow Jun-ichi Nakayama and colleagues provide the first in vivo evidence that methylation of histone H3 plays a central role in the transcriptional repression and organization of large chromosomal domains. Their work also defined a highly conserved pathway wherein histone deacetylases and histone methyltransferases act cooperatively to establish a “histone code essential for higher order of chromatin assembly
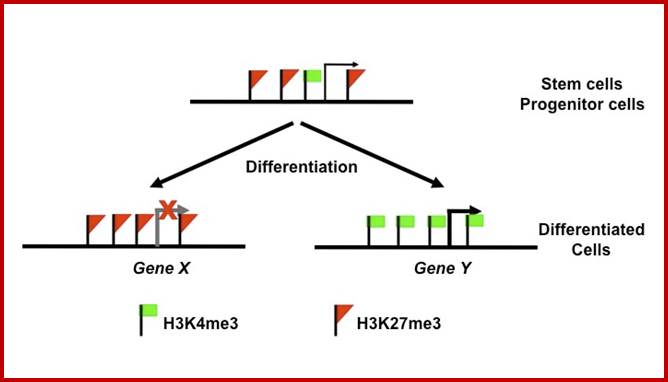
Figure; Both H3K4me3 and H3K27me3 marks are critical for the maintenance and differentiation of stem cells. H3K4me3 is the epigenetic mark for transcriptionally active chromatin, and H3K27me3 is the epigenetic mark for transcriptionally inactive chromatin. In stem cells and progenitor cells, many genes important for developmental control are marked by both H3K4me3 and H3K27me3 marks and therefore are poised for transcription activation or repression. Thus, the enzymes that regulate the levels of H3K4me3 and H3K27me3 are important for the homeostasis of the stem cells.
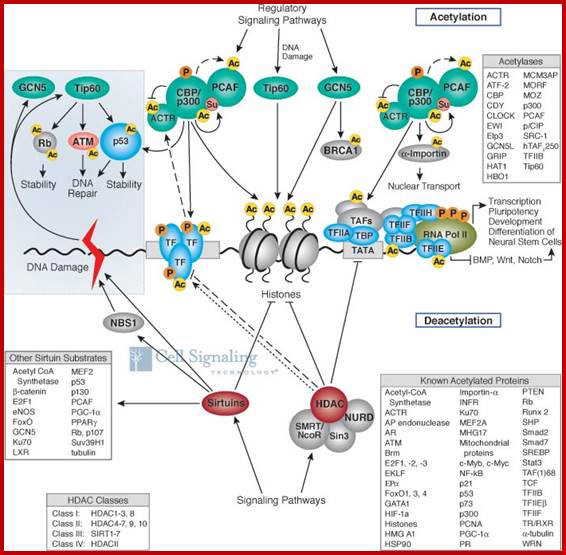
Pathway Description of acetylation and deacetylation: Result activation and deactivation of Genes in specific
Protein acetylation plays a crucial role in regulating chromatin structure and transcriptional activity. Many transcriptional co activators possess intrinsic acetylase activity, while transcriptional co repressors are associated with deacetylase activity. Acetylation complexes (such as CBP/p300 and PCAF) or deacetylation complexes (such as Sin3, NuRD, NcoR and SMRT) are recruited to DNA-bound transcription factors (TFs) in response to signaling pathways. Histone hyper acetylation by histone acetyltransferases (HATs, also called KATs for lysine acetyltransferases) is associated with transcriptional activation, whereas histone deacetylation by histone deacetylases (HDACs or KDACs) is associated with transcriptional repression. Histone acetylation stimulates transcription by remodeling higher order chromatin structure, weakening histone-DNA interactions, and providing binding sites for transcriptional activation complexes containing proteins that possess bromodomain, which bind acetylated lysine. Histone deacetylation represses transcription through an inverse mechanism involving the assembly of compact higher order chromatin and the exclusion of bromodomain-containing transcription activation complexes. Histone hyper acetylation is a hallmark of silent heterochromatin. Site-specific acetylation of a growing number of non-histone proteins, including p53 and E2F, has been shown to regulate their activity, localization, specific interactions as well as stability/degradation, therefore controlling a variety of cellular processes, such as transcription, proliferation, apoptosis and differentiation. It is becoming clear that crosstalk between acetylation and other modifications, such as methylation and phosphorylation in histone and non-histone proteins, plays a critical role in the final output signals. Some modifications are required in a combinatorial order to exert an effect, while others are mutually exclusive. In this context, the different putative combinations likely work to diversify cellular signaling networks, providing a much broader way to modulate responses. At an organismal level, acetylation plays an important role in immunity, circadian rhythm and memory formation. Protein acetylation is becoming a favorable target in drug design for numerous disease conditions. However, most of the available activators and inhibitors are as yet broad and non-specific. Recent studies have provided detailed analysis on the function and structure of the different KATs and KDACs, therefore we could expect the development of better and more specific modulators in the near future.
Dynamics and Interplay of Nuclear Architecture, Genome Organization, and Gene Expression:

Transcriptional regulation in human cells is a complex process involving a multitude of regulatory elements encoded by the genome. Recent studies have shown that distinct chromatin signatures mark a variety of functional genomic elements and those subtle variations of these signatures mark elements with different functions. To identify novel chromatin signatures in the human genome, we apply a de novo pattern-finding algorithm to genome-wide maps of histone modifications. We recover previously known chromatin signatures associated with promoters and enhancers. We also observe several chromatin signatures with strong enrichment of H3K36me3 marking exons. Closer examination reveals that H3K36me3 is found on well-positioned nucleosomes at exon 5′ ends, and that this modification is a global mark of exon expression that also correlates with alternative splicing. Additionally, we observe strong enrichment of H2BK5me1 and H4K20me1 at highly expressed exons near the 5′ end, in contrast to the opposite distribution of H3K36me3-marked exons. Finally, we also recover frequently occurring chromatin signatures displaying enrichment of repressive histone modifications. These signatures mark distinct repeat sequences and are associated with distinct modes of gene repression. Together, these results highlight the rich information embedded in the human epigenome and underscore its value in studying gene regulation.
Indeed, at promoters, we identified a number of distinct signatures, each associated with a different class of expressed and functional genes. We also observed several unexpected signatures marking exons that directly correlate with the expression of exons. Finally, we recovered many places marked by two distinct repressive modifications, and showed that they mark distinct populations of repetitive elements associated with distinct modes of gene repression. Together, these results highlight the rich information embedded in the human epigenome and underscore its value in studying gene regulation.
Open chromatin encoded in DNA sequence is the signature of ‘master’ replication origins in human cells
Benjamin Audit1,2, Lamia Zaghloul1,2, Cédric Vaillant1,2, Guillaume Chevereau1,2,Yves d'Aubenton-Carafa3, Claude Thermes3 and Alain Arneodo1,2
For years, progress in elucidating the mechanisms underlying replication initiation and its coupling to transcriptional activities and to local chromatin structure has been hampered by the small number (approximately 30) of well-established origins in the human genome and more generally in mammalian genomes. Recent in silico studies of compositional strand asymmetries revealed a high level of organization of human genes around 1000 putative replication origins. Here, by comparing with recently experimentally identified replication origins, we provide further support that these putative origins are active in vivo. We show that regions ∼300-kb wide surrounding most of these putative replication origins that replicate early in the S phase are hypersensitive to DNase I cleavage, hypo methylated and present a significant enrichment in genomic energy barriers that impair nucleosome formation (nucleosome-free regions). This suggests that these putative replication origins are specified by an open chromatin structure favored by the DNA sequence. We discuss how this distinctive attribute makes these origins, further qualified as ‘master’ replication origins, privileged loci for future research to decipher the human spatio-temporal replication program. Finally, we argue that these ‘master’ origins are likely to play a key role in genome dynamics during evolution and in pathological situations.
Cell cycle regulation of chromatin at an origin of DNA replication
Jing Zhou, Charles M Chau, Zhong Deng, Ramin Shiekhattar, Mark-Peter Spindler, Aloys Schepers and Paul M Lieberman

Model of cell cycle coordinated histone tail modification and chromatin remodeling at OriP. An SNF2h/HDAC2 complex can be colocalized to DS during G1, simultaneously with histone H3 deacetylation, chromatin remodeling, and MCM loading.
Chromatin signature at Origin:
Model of cell cycle
coordinated histone tail modification and chromatin remodeling at OriP.
An SNF2h/HDAC2 complex can be co localized to DS during G1, simultaneously with
histone H3 deacetylation, chromatin remodeling, and MCM loading.
Model of cell cycle coordinated histone tail modification and chromatin
remodeling at OriP. An SNF2h/HDAC2 complex can be colocalized to DS
during G1, simultaneously with histone H3 deacetylation, chromatin remodeling,
and MCM loading.
Model of cell cycle coordinated histone tail modification and chromatin
remodeling at OriP. An SNF2h/HDAC2 complex can be colocalized to DS
during G1, simultaneously with histone H3 deacetylation, chromatin remodeling,
and MCM loading.
M9 - Control of DNA replication by epigenetic modification and chromatin remodeling
To ensure genome stability, DNA replication is tightly controlled at its initiation stage and strictly limited to once-per-cell-cycle. Replication competence is gained only in the G1 phase of the cell cycle, a process called licensing, which is characterized by the formation of pre-replicative complexes (pre-RC). Pre-RCs are formed at specific sites called replication origins. Little is known how pre-RC assembly relates to the chromosomal context, and whether chromatin remodeling and posttranslational modification (PTM) are involved in this process.
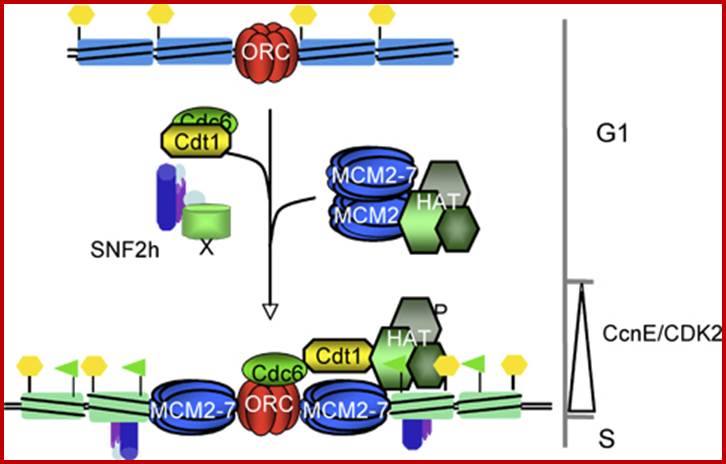
Model of chromatin remodelling complexes and histone modifying enzymes at replication origins. Both enzymatic functions may influence pre-RC assembly at replication origins and origin selection in the context of chromatin.
It is the aim of this proposal to study two aspects of the relationship between pre-RC formation at replication origins and in their chromatin environment. (i) Does pre-RC assembly require the action of a specific chromatin-remodeling complex? (ii) Is pre-RC formation during G1 associated with specific histone modifications? To address the first question, we will use a proteomic approach to identify pre-RC components associated with a SNF2h-containing remodeling complex. Using ChIP-on-Chip we will determine, whether all origins or a specific subset of them requires the activity of chromatin remodeling complexes. The second aspect focuses on the role of a specific histone acetyltransferase, Hbo1, and its cofactor ING5 and, on how the activity of this complex is integrated into cell cycle regulation of pre-RC formation.
Predictive chromatin signatures in the mammalian genome
Gary C. Hon1,2, R. David Hawkins2 and Bing Ren1,

Modification of histone H3.
Lysine residues on histone H3 can be mono-, di- or tri-methylated. Shown are modifications H3K4me1, H3K4me3 and H3K36me3, which mark:
1. H3K4me1active/poised enhancers,
2. H3K4me3 active/poised promoters,
3. H3K36me3actively transcribed regions, respectively. Me = methylation.
HMG-14 and Transcriptional Elongation
The nucleosomes that did remain appeared to be devoid of histone H1 and to contain instead nonhistone chromatin proteins HMG-14 and 17 proteins. HMG-14 is able to stimulate the elongation phase of transcription by RNA polymerase II. It does not promote the initiation of transcription and it does not affect transcription elongation on nucleosome-free DNA. It appears that the association of HMG-14 with nucleosomes is part of the process that maintains transcription on nucleosomes.
The recent findings from these studies reaffirm the idea that unique chromatin signatures correlate with cell type and function, and that a given chromatin state is a balance of multiple epigenetic marks and implies that cell fate can be experimentally manipulated through tipping this balance.
Heat shock protein chromatin signature:
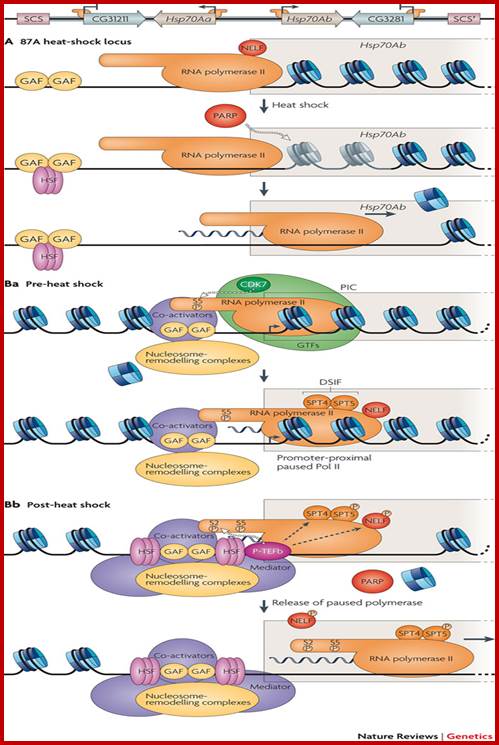
a | At the top is a schematic of the Drosophila melanogaster 87A heat-shock locus; the example of Hsp70Ab is used below. Equivalent events occur at Hsp70Aa. The arrows at the Hsp70Aa and Hsp70Ab promoters indicate the direction of transcription through the gene. RNA polymerase II (Pol II, shown in orange) and GAGA factor (GAF or Trithorax-like) are bound at the promoters of the Hsp70 genes before heat shock.
After heat shock, the transcriptional activator, heat-shock factor (HSF), forms a stable trimeric complex that binds the Hsp70 promoter105, 106. Heat shock stimulates nucleosome loss at the Hsp70 locus. Nucleosome loss is dependent on HSF, GAF and poly(ADP)-ribose polymerase (PARP)64. PARP localizes at many sites along polytene chromosomes but only catalyzes formation of ADP-ribose polymers from donor NAD+ at the heat-shock loci after induction by heat shock107.
Nucleosome loss proceeds outwards from the 5′ end of the Hsp70 genes ahead of Pol II, extending as far as the SCS and SCS′ boundary elements (region of loss is grey in top panel). The CG31211 and CG3281 genes are not transcribed (as shown by blunt-headed arrows at their promoters) although Pol II is bound at their promoters and nucleosomes are lost.
Ba. Before heat-shock activation at Hsp70, GAF recruits co-activators, the GTFs and ATP-dependent nucleosome-remodeling complexes, thereby facilitating pre-initiation complex (PIC) formation at the promoter. At Hsp70, however, PIC formation is not sufficient to activate productive transcription elongation. The CDK7 subunit of TFIIH phosphorylates serine 5 (S5) of the carboxyl-terminal domain (CTD) and Pol II initiates transcription into the first 20–40 bases of the gene, at which it pauses at the promoter-proximal pause site. Pol II is held here by the negative elongation factor (NELF) and DRB sensitivity-inducing factor (DSIF), which is composed of SPT4 and 5.
Bb. Heat shock induces binding of HSF, which recruits additional co-activators and nucleosome-remodeling activities. HSF is required but is not sufficient to recruit the pause release factor P-TEFb to Hsp70. Recruitment of Mediator by HSF, which occurs independently of PIC formation, might contribute to P-TEFb binding. Recruited P-TEFb phosphorylates S2 of the CTD, SPT5 and NELF. These phosphorylation cause NELF to dissociate from Pol II, releasing polymerase into productive transcription elongation. Although Pol II still pauses briefly at the promoter-proximal pause site under heat-shock conditions, the duration of these pauses are much shorter than at normal temperature.
Genetic Imprinting:
Taking human species as an example, sperms contain either one X or one Y chromosome. The X-chromosome is genetically active and Y-chromosome mostly inactive with the exception of few genes which are expressed in males.
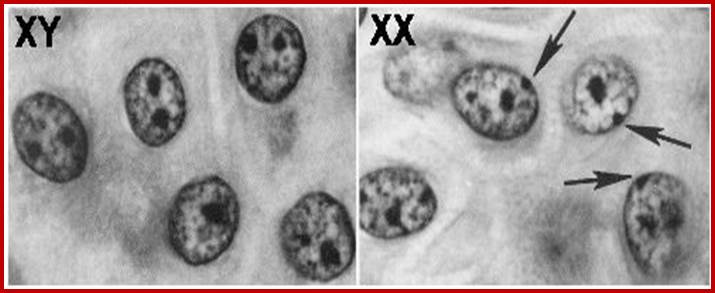
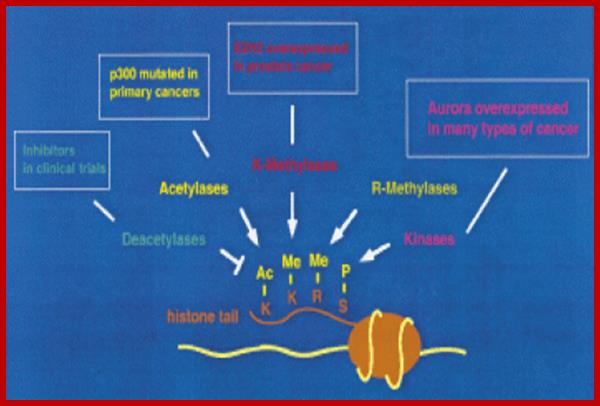
In females, early in embryonic stage both X-chromosomes from Ma and Pa inherited, are active. But as cell division continues and when cell differentiation initiates, at this point of time, in some tissues X from Pa becomes inactive and the chromosome from Ma remain active. In some other tissues the chromosome from Pa is active and chromosome from Ma remains inactivated. So the females have mosaic of active and inactive X chromosomes in different tissues. Allelic gene expression in such tissue expresses epigenetic variation. The X-chromosome that is inactivated when stained appears a dark body is called Bar body. The X-chromosomes carry several hundreds of genes and many of them are allelic.
Differentially, one of the two X chromosome in one tissue and the other, in other tissue, becomes inactive, which is attributed directly X inactivation transcript. The gene for such transcript is Xic. The Xic gene produces Xist RNAs. If one of the chromosome inactive the other X chromosomes remains active for another transcript acts as antisense RNA against Xist RNA.
Thus an allele with one dominant and the other recessive, in one tissue the dominant gene is expressed and in another tissue the recessive character is manifested, which phenomenon is called genetic mosaics, or what is popularly called calico-women. In another tissue it can reverse expression. A good example is of coat colors in cats and other animals. This is often referred to as epigenetic phenomenon.
Silencing of certain allelic genes in one tissue and expression of the other allele in another tissue is very common, one such gene allelic pairs of genes is IGF-II (insulin like growth factor-II). It is an example for genetic imprinting. Molecular basis of development poses a situation for classical biologists what is determination and what differentiation is and how to differentiate between the two.
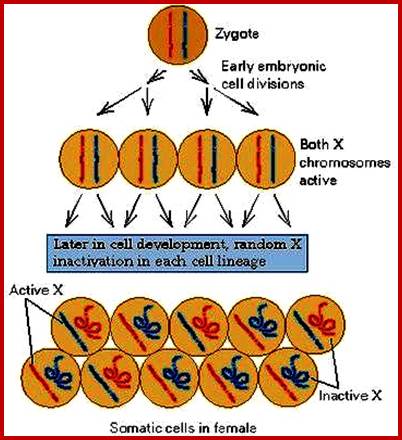
Determination in molecular term is the availability of certain specific factors and assembly of the same on to a set of promoters for transcription, which ultimately determine the fate of the said cell. Inducing the expression of certain genes in a cell which changes the character of the cell and it will propagate so, is termed as determination.
Once the gene inducing factors are in place, expression of the genes, which are different from the earlier set, determines what is the phenotype or the character of the cell, which is different from the previous state; such cell or cells are called differentiated.
000000000000000000000000000000000000000000000000000000000000000000000000