Repair Mechanisms:
Co-Replication Repair Mechanisms:
In general, most of the replication errors in E.coli are repaired during replication itself, by the epsilon subunit of DNA pol-III; and by DNA pol-I.
· The subunit epsilon of DNA pol-III, which has 3’->5’ exonuclease property, by backward sliding, removes any wrong nucleotide incorporated during replication, which is the single most important error proof repair enzyme involved in DNA replication. It is the most efficient repair mechanism; replication error rate is one in 1o^6 to 10^8 nucleotides incorporated. The rate with which it replicates is 40000 to 60000ntds per minute (amazing).
The role of DNA pol-I is not just restricted to replication stage, it can perform repair function whenever and wherever such repairs required, and this includes recombination processes. If any mismatches remain not repaired by DNA pol Holozyme, then DNAP I performs the process.
· In an experiment, a poly-(A) template, when primed with oligo-Ts with one C at its 3’end is provided, the DNA pol-I first remove the ‘C’ from the oligos and then extend the chain by its polymerase function in reverse direction, a parexellent repair process.
3’AAAAAAAAAAAAA5’
5’TTTTTTC>3’
3’AAAAAAAAAAAAA5’
5’TTTTTT>3’
(-) C is removed
3’AAAAAAATTTTTtTT5’
5’TTTTTTTTAAAAAAA>3’
Oligo T primer is extended
The above feature of DNA pol-I shows if there are any mismatches during incorporation of nucleotides, if they are not removed by epsilon unit, then Pol-I will do it.
· The ligation is performed by the respective Ligases,

DNA Pol-I during repair removes any non-complementary 3’base, and then it uses its polymerase activity to fill the gap; http://www.web-books.com
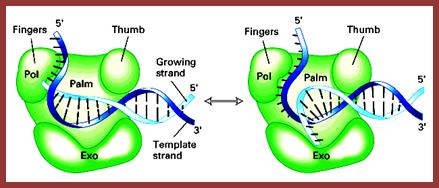
This a DNA Pol I model; http://slideplayer.com
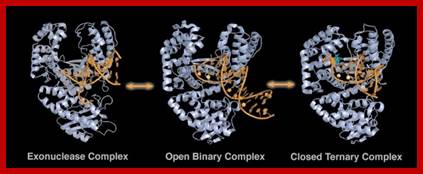
Replicating and editing; http://www.scripps.edu
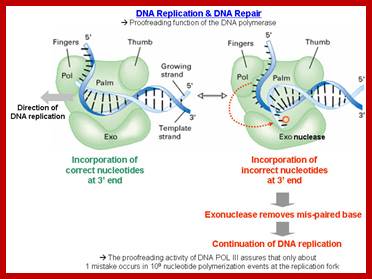
http://classroom.sdmesa.edu
But DNA pol II from bacteria is exclusively meant for repairing the DNA damage, but not much is known (?).
Mis-Match Repair mechanism in newly formed DNA strands:
DNA replication is not foolproof process. In spite of the presence of proof reading mechanism, occasionally some errors such as mismatch do occur and they are left as defects, but biosystems are endowed with post replication repair mechanism to remove mismatched nucleotides. It is not only mismatched bases, there may be damages to DNA as base modifications with adducts and even dimers. Mismatch repair system is more effective in Transversions types than transition type mismatches. Mismatch repair requires a number of gene products such as Mut-S, Mut-H, Mut-L, Mut-U (= Uvr-D).
Mut-S = 97kd,
Mut-D = dna-R (it is subunit of DNA pol-III),
Mut-L = 70kd,
Mut-H = 25kd, it is an endonuclease,
Mut-U = Uvr-D = helicase-II,
Exonuclease-I
Mut-L and S remove wrong base pairings of C = T,
Mut-Y, remove mismatch pairing between A = G and A=C
This process requires a specific sequence in the DNA, such as GATC. Generally, the ‘A’ of GATC is methylated by SAM, Adenosyl methyl transferase.
In bacterial systems replication machinery uses this sequence where methylation of GATC in both strands (full methylation) is required for initiation of replication; in some cases methylation of the said sequence is absolutely required for transcriptional activation, ex. Activation of dna-A gene in E.coli and initiation DNA replication at specific Ori sites
On replication, one of the parental strands retains methylation and the new strand is yet to be methylated. If there any such mismatch errors, the repair system requires such hemi-methylated regions to differentiate between the parental and newly formed strands, for executing the process.
· Mut-S binds to DNA and scans for the lesion, when lesion is found, it binds. The Mut-L joins Mut-S complex. These proteins are lesion identifiers.
At this point another protein called Mut-H joins and locates hemi-methylated GATC whose location can be 100 or more nucleotides away from the site of lesion. If the GATC is fully methylated, the protein does not recognize the sequence.

ATR signalling; Edward A. Nam, David CortezMut S, MutL, MutL mediated repair. http://www.biochemj.org
This diagrams shows the homologues of MutSL called MutS a or MutS b in eukaryotes along with PCNA associate with mispaired region of the post-replicated DNA and perform the repair process similar to E.coli.
· After binding, by protein–protein interaction Mut-L/Mut-S associate with Mut-H, this interaction leads to the pulling of the DNA till they reach GATC sequence, this generates a loop containing the lesion.
At the same time this interaction activates Mut-H, which has endonuclease activity, which produces a nick adjacent to GATC in the new strand. If the lesion is towards 5’ end of the GATC it produces the nick towards the 5’ end of the GATC in the new strand.
· The Mut-U a helicase (helicase-II) and an exonuclease-I removes the nicked strand including the lesion region and beyond the lesions.
If the lesion is located towards 3’end of the GATC of the new strand, the nick is made towards 3’ end of the GATC and exonuclease-I is replaced by Rec-J, an exonuclease, which has both 5’—3’ and 3’-5’ activity, thus it removes the cut strand.
· Exonuclease III can substitute Rec-j.
The gaps are filled by DNA pol-I and ligated by DNA ligase.
· Human systems also contain Mut genes except that they lack Mut-H.
Mut-L prevents mis-match of promiscuous genetic recombination between poorly matched DNA.
Certain proteins in E.coli prevent such poorly matched recombination. These enzymes not only remove mismatched base pairs, but also correct small deletions and additions found in the new strand.
Mut T, M, Y system:
Mut-T:
Whenever dGTP as a nucleotide undergoes oxidative damage to generate 8-oxy-GTP; this has a severe effect for it can base pair with Adenine base. Thus the changed nucleotide is not available for incorporation into DNA.
Mut-M
When the base pair G-C, where G is subjected oxide (^-) radical reaction, it can be converted to 8-o-G; this happens in vivo all the time in the cell by oxidative reactions. In such situations the Mut-M removes 8-O-G, so it will be left with C alone partner, and the template with C is used to complement with G to generate G-C pairing.
Mut Y:
The G always paired with C. Whenever 8-O-G gets oxygenated, it pairs with A; in such situations Mut-Y removes A, leaving 8-O-G without its partner, then in the next replication event it pairs with C, but repair is done by Mut M, where it removes 8’O=G left with C alone, which is base paired by G, thus restores.
Note: Mut M and Mut Y are actually a kind of glycosylases, which remove unusual bases when found in the normal DNA.
Repair of altered bases or missing bases in the existing DNA:
Deamination of cytosine is also a common and every day process. Deamination of cytosine produces Uracil in its place. This uracil is removed by Deoxy Uracil glycosylase, which in turn causes Apyrimidinic site.
Deamination and oxidation of 5-methyl cytosine results in Thymidine. Similarly deamination of adenine and guanine results in the formation of Hypoxanthine and Xanthine respectively. Such changes cause mismatch base pairing. Such bases are removed base excision mechanism where specific DNA glycosylases remove the damaged bases, leaving a hole in the DNA strand.
In all the above cases and also if there are any cyclobutane dimer structures the enzymes bind to the affected site and flip base towards the enzyme active site and perform the glycosylase reactions.
Whenever there are perturbations such as increased acidity or increased heat, makes the glycosidic bonds (very weak bonds) between bases and the deoxy sugar to break. This happens every day in every cellular genome. Hydrolysis of the glycosidic bond creates an APu or APy (Apurinic and Apyriminidic) sites.

http://flylib.com

Removal the base and excision of the phosphodiester bond and filing up the gap;
Base excision and repair; http://www.acsu.buffalo.edu
· In bacteria the gene to repair this damage is called as Ung^+, its mutant is Ung^-. In Ung^- mutants, if Deoxy-Uracil nucleotides are incorporated, by mistake, Uracil remain in the new strand and it is not removed and results in error and mutations in the second generation or if this template is used for transcription. This takes place generally in lagging strand. Ung glycosylases are always associated with replisomes. But action of C-glycosylases is 100 times faster on single strands than double stranded DNA.
· In humans there are four such Uracil glycosylases. Human hS MUG-1 remove any such Uracils from both single and double stranded DNAs. This activity is performed either at replication time or transcription process. The other human Ungs are TDGs, MBD4, they remove either Us or Ts if they are paired with Gs.
In experiments to create general mutations in a plasmid DNA, bacteria cells with Ung^- strains containing plasmids are used, for they are lacking in deoxy uracil glycosidase. When such cells are fed with deoxy-Uridine nucleotides with low concentrations, so that one such nucleotide is incorporated in one thousand or more, point mutations thus obtained can be screened. This is also a method to create point mutations to see what will be its biochemical or phenotypic effects.
· In base excision repair mechanism, each species of glycosylases recognize an altered base and remove them by hydrolytic cleavage of the respective glycosidic bond. There are six or more types of glycosylases: 1) Remove deaminated A s, 2) remove alkylated bases such as 7-methyl Guanines or 3- methyl Adenines, 3) Remove oxidized bases such as 8-hydroxy Guanines, 4) Remove bases with open rings, 5) Convert bases where C = C double bonds to C-C single bonds and 6) Remove Uracil, some recognize T-T dimers.
If 5’methylated Cytosine is deaminated, it generates a Thymidine, which is not removed by any glycosylases and it remains as an error.
· Methylation of Guanine at 6’-O position can cause automatic breakdown of glycosidic bond and generation of an Apurinic site.
There are other glycosylases, which can also remove methylated Adenine, Cytosine and Thymidines, causing both Apurinic and Apyrimidinic sites.
There is another set of enzymes called AP lyases which act on Deoxyribose sugars and open the sugar rings, as a consequence the nucleotide at its 3’ end is released.
· Such sites with missing bases are undesirable and such lesions are repaired to normalcy by a set of enzymes.
Apurination and Apyriminidation produce a hole in the strand, yet the DNA backbone retains the S-p-S-p-S backbone intact. Such lesions are removed by breaking the S-p-S bonds by AP endonucleases. When the nick is produced, the same can be expanded by exonucleases and DNA pol-I fills the gap and DNA ligase seals the nick. The most important polymerase involved in DNA repair mechanism is DNA pol-b, it is responsible for filling the gaps.

In above illustration AP lyase opens the AP sugar, in this process the nucleotide next to it is released; http://www.acsu.buffalo.edu/

Uracil N-glycosylase-A base excissionm repair enzyme, Allen Gathman by https://www.flickr.com
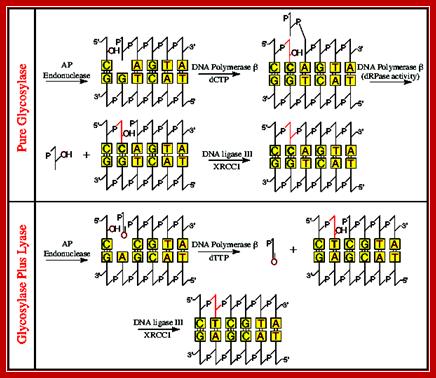
The above line diagram illustrates how AP hole in ds DNA are removed by the combined action of glycosylases and Lyases; the fill up is performed by DNA Pol beta which is specific to DNA repair. DNA repair overview, Chen Yonggang, Zhejiang Univ. https://www.slideshare.net

An intron encoded enzyme; http://www.wenmr.eu
· Class-IIAP endonucleases recognize such lesions, which scan the strands and locate the gaps or lesions, and nick the damaged strand at Apurinic or Apyrimidinic sites at the 5’ end of the lesion.
This gap or the nick is recognized by DNA pol-II beta, and it removes nucleotides not only one but up to ten or more nucleotides by its 3’exonuclease activity and then by its 5’-3’ polymerase activity fills the gap. Then the ligases seals with a phosphodiester bond between the 5’p of one nucleotide and 3’ OH group of the other nucleotide, by covalent bond formation.
· Class-I has both glycosylases and endonuclease function.
Class III AP-endonucleases also performs Nucleotide Excision Repair (NER) of missing bases. This enzyme has dual functions, for it has a domain, which is involved in the removal glycosidic bonds of altered or damaged bases (including oxidized bases). Another domain has the function to make nicks in P-S-P backbone next to the missing base. The enzyme is a monomeric protein with 211 amino acids and contains 4Fe-4S clusters; this character is conserved among the members of other bacteria, yeast system and mammalian systems.
Note:
Class–I AP endonucleases have both N-glycosylase, as well as AP endonuclease activity.
Class-II endonucleases make incisions next to AP sites.
Exonuclease III and exonuclease-IV remove fragmented strands and they are bacterial enzymes. T4 phage produces an endonuclease, which removes pyrimidine dimers.
Nucleotide Excision Repairs Mechanism:
General; can be intra-strand or Inter-strand:
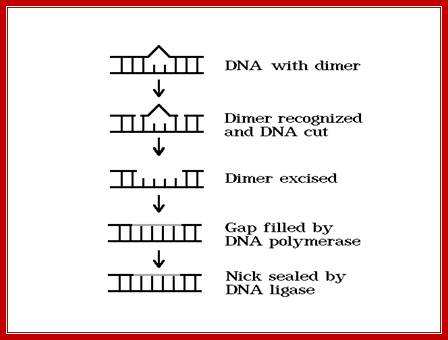
Cut and remove the nucleotides at the site of DNA damage; http://www.phys.ksu.edu/
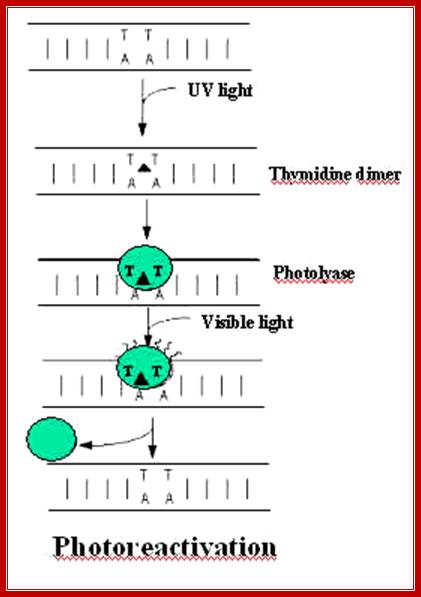
Thymine dimer, illustrating the cyclobutyl ring formed (blue) which alters the DNA structure;Trishul;http://www.bath.ac.uk/; http://trishul.sci.gu.edu.au/

The above diagram shows stepwise process by
which T-T dimers repaired to normalcy; https://www.slideshare.net/
If the lesion are large they distort DNA helical structure, and such distortions are removed by a method called Nucleotide excision repair. In E.coli the repair enzyme is multisubunits complex. In eukaryotic system nearly 16 subunits complex is involved in nucleotide excision process.
The above diagram explains the process by which the TT dimers or any other intra strand dimers are removed.
E.coli the key enzyme complex is called uvrABC-Excinuclease. It consists of three subunits called Uvr-A (104Kd), Uvr-B (378kd) and Uvr-C (68kd). UvrA is a dimer complexes with UvrB (A2B) associates with DNA and scans it and halts at the damaged site.
During transcription, the protein called Mfd first identifies the damaged site and displaces RNAP,then directs the UvrABC system to the damaged site on the template strand. It can locate pyrimidine dimers or any such dimers in DNA under normal situations. Not only the dimers, it also recognizes heavy adducts.
How it recognizes and what is the basis is yet to be discerned. Once the site is recognized, UvrA dissociates and Uvr C associates with Uvr B. The Uvr B (having endonuclease property) cuts at 3’ end of the damage at 3-4 ntds from the damaged site. And Uvr C cuts at 5’ site 7th nucleotide from the damaged site. Then another subunit called Uvr D joins, which is an helicase, removes the cut segment. The gap, however is filled by DNA pol-I, DNA pol II and DNA pol III and ligase seals the 3’OH and 5’P ends of the nick into a Phosphodiester bond. However it is worth remembering that the extent of the excision can be 12 ntds, or 1500 to 9000 ntds.
This is a general pathway by which they remove the lesions including cyclobutane derived T-T dimers. They are also very effective in removing Benzo (alpha) Pyrine–guanine (due to tarring effect of cigarette smoking). This Excinuclease activity is very novel system.
In the case of T4 endonuclease-V enzyme, called pyrimidine glycosylase (T4pdg). The enzyme once locates the lesion site such as T-T dimer; it flips out adenine against T-T, then it get access to the T-T dimer and removes.
In eukaryotic systems the Excinuclease complex consists of 16 subunits and the repair process is more or less similar to E.coli but linked to the repair of transcriptional errors.
Repair of DNA with Dimers or bulky adducts in new strands:
Removal of adducts like Thymidine dimers:
Thymidine dimerization is actuated by UV radiation at 260-300nm. Such dimers, if not corrected, prevent normal replication. Such dimers can create deletions in the next round of replication, which can have devastating effect on cellular functions.
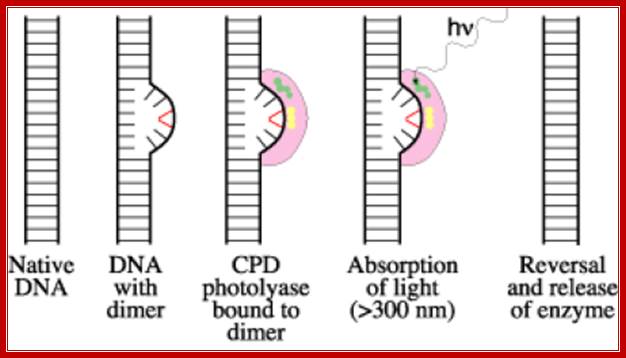
The diagram be shows how the Photolyase uses light energy and transfer the light energy to Cyclobutane rings to open up to generate individual nucleotide free to base pair with their normal bases. Before they execute repair functions they flip the dimers towards enzyme surface, then perform reaction. www.acsu.buffalo.edu
· Photo-reactivation method involves Photolyases, which are inducible enzymes by any damage to DNA, are involved in removal of thymidine dimers or thymidine-cytosine dimers by a process called photo-reactivation mechanism. These are also called deoxy ribo- pyrimidine Photolyase (DRPP).
The Photolyase protein is a monomer with a Mol.wt of 55-65kd. The enzyme has a chromophore N5-N10 methyl tetra hydrofolate or it is also called 5’ deaza flavin. The cofactor for this enzyme is FAD. This enzyme binds to DNA and scans for the lesions and when finds a lesion it tightly binds. When such enzymes are exposed to 300-600nm light, they get activated. The enzyme flips the dimers into its active site. The activated enzyme transfers electrons to T-T dimers to open the cyclobutane ring and renders them as monomers.
Interstrand Excision Repair:
Interstrand cross-linking is produced by psoralin reactions, Mitomycin-C actions and sulfur mustard reactions.
· In such conditions, repair involves the cutting of both the strands and replacing both strands by trans-splicing from the intact ds DNA, which have such sequences.
This is possible only when another normal copy of intact DNA is also found in the same cell. If E.coli cells are dividing exponentially, more than one copy of DNAs are found in the same cell. In such cells, if the damage occurs because interstrand cross-linking, and then the normal and correct copy can be used for replacing the Interstrand lesion.
· This is achieved by excision and recombination. This requires the participation Uvr-ABC excinucleases and also Rec-A and Rec-BCD participation. It requires homologous pairing between the intact DNA strands and damaged strands to generate a triple stranded structure. Uvr-ABC nuclease produce nicks on either side of the damage. Another nick is made in one of the strands in the undamaged DNA. One of the ends is threaded into the damaged DNA to generate triple stranded structure.
Then the Uvr-ABC nicks in the other strand of the damaged DNA, thus the entire section of the damaged region is removed.
· But the cut end of the normal DNA is now already threaded into the damaged DNA, which provides substrate for Rec-A and Rec-BCD mediated recombination, which not only corrects the damaged DNA, but also repairs one of the nicked strand from normal DNA.
In these sequential and concerted reactions, DNA pol-I and ligases perform gap filling and ligation functions.
Required enzyme components in E.coli:
Error Prone Repair-Translesion recombination repair:
· Uvr-ABC exonuclease components.
· Rec-A recombinase, Mol. Wt of Rec-A is 38 KD, it has strand exchange and endonuclease activity, it is also responsible for homologous paring of DNA strands.
· Rec-BCD nuclease (rec-BC subunits have exonuclease activity), Rec-B and Rec-C together has a Mol.wt of 300kd. Both have unwinding and nuclease functions. They bind to the exposed free end of DNA.
· Rec-F.
· UmuC and umuD,
· RecA.
· RecBCD.
· DNA pol – I.
· DNA ligase.
· In eukaryotic systems diploids and triploid provide the required normal complement DNAs for they have homologous chromosomes.
Some damages are severe, like Mitomycin-C induced interstrand cross linking, 4’ to 5’ interstrand dimerization or addition of strong adducts or loss of bases from single stranded genomic DNA, are few of the damages difficult to repair. Even though repair is performed, the repair is always prone to errors.
· For example phi X174 ssDNA genome, when suffers from the loss of one or two bases, the host cell has no complementary strands to use recombination repair system. In such cases the cell takes some desperate steps to repair or salvage, as the last resort; such repair system is always prone to errors.
Existence of such error prone repair mechanism has come to light by the discovery of two mutants called Umu-C and Umu-D; here Umu stands for immutable to UV radiation; umuCD code for DNA pol-V.
· These are SOS genes; respond to DNA damage, whether it is in the form of nucleotide dimers or single strand breaks, such damages act as signals for the response genes to get activated. Cells, mutant for Umu-C and Umu-D are highly sensitive to UV damages.
Rec-A is also required for these response genes to act. Rec A activates umuD. The umuC and D operons are induced by DNA damage and RecA activates them for umuD self cleavage similar to LexA. UV can cause damage to DNA, cross linking of bases and alkylation of bases all these act as stimuli for SOS response which can lead to excision repair or recombination repair. Response to the signal is very fast.
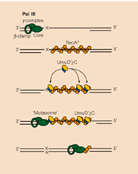
If a lesion like nucleotide dimers left without repair, in the next round of replication, when DNA pol-III encounters such lesions, it halts a while and skips the lesion region by about hundred or more nucleotides, then it continues replication in a normal way. In this process another the partner strand is replicated normally. So this ds stranded daughter DNA has the correct information.
The gap left in the damaged strand is filled by Rec-A and Rec-BCD mediated recombination process using the newly formed sister ds DNA.
The gap can also be repaired by another group of proteins called umuC and umuD. They are induced by damages to DNA and also if replication fork stops they are activated. These two as D2C complex join the DNA that with the gap and replicates the complementary strand and places two As against T_T dimers and progresses. Such translesion repair is error prone. Such types of translesion require DNA pol IV also. DNA pol IV and polV are a part of the family of translesion polymerase found in all organisms.
Protein D2C binds to RecA bound single stranded DNA, at the same time it gets associated with DNApol III with assistance beta clamp. This complex is called mutasome and repairs, of course with errors.
DNA pol h is TLS polymerase found in all eukaryotic systems. They lack fidelity and processivity, thus they cause one error for every 1000 nucleotides incorporated. DNA pol-h places two A residues against T-T dimers and continues. Stalled replication due to heavy adducts or T-T dimers, or break in one of the strands, repair is done by recombination then switch back to normal course of replication. However this type of replication is origin independent mode of replication.
Such process has been observed while investigating phiX174 DNA replication. This requires PriA, priB, priC, DnaB, DnaC, DnaG and DnaT. Such primosome assembly is called Restart primosome complex. It requires DNA pol II which eventually gives way to DNA pol III. Recombination is required wherever replication leaves a gap or if there is break in the replicating strand. Inspite of it if any lesions are left they are removed by base excision or nucleotide excision.
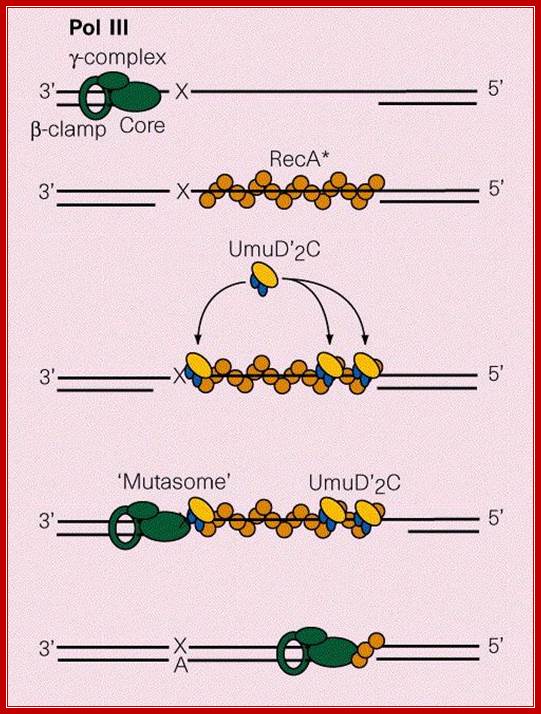
Mutasome, explained in the text is involved in error prone recombination repair. Myron F. Goodman;https://www.nature.com
Mammalian Photolyases:
In mammals, especially humans, Xeroderma pigmentosa is a skin disease cause by a defect in Photolyase (Phr) enzyme systems. Phr genes code for them. When such patients are exposed to bright light for longer duration they are susceptible for developing blisters and skin cancer. In human systems out of 11 genes involved DNA repair six of them are involved in repair of Xeroderma pigmentosa.
· In yeast RAD3 is one of the 11 genes; they have homologues in human system.
In yeast cells, ten radiosensitive RAD genes have been identified. Six of them are involved in recognizing the damaged DNA regions.
In human system, single cell line cultures of Chinese hamsters have revealed, the existence of DNA repair system for Mitomycin induced damage and UV mediated damage and other related damages.
· In human system excinucleases are found to exist and contain about 16-17 polypeptides and some of them are associate with certain Transcriptional factors such as TFII-H complexes. There are two TFII-H systems one for transcriptional initiation-elongation and the other for repair of translesion in the template strand involved in transcription
Xp-A: damage recognition,
Xp-B: helicase, a subunit of TF II-H,
Xp-C ?
Xp-D: pleotropic protein, has helicase activity, stabilizes TF II-H,
Xp-E
Xp-F: Nuclease belongs to TF II-H (TFII-H consists of 8 or more subunits),
Xp-G: Nuclease,
Xp-V = yeast RAD3D
ERCC-1: It is the part of nuclease belonging to Xp-F and also Replicase protein called Rp-A. HSSB binds to Xp-F-ERCC-1 with Xp-A binds to lesion site (HSSB is a RP-A protein).
Mutations in XP components makes fibroblast cells fail to repair UV induced damages or damages due to bulky adducts.
A number of XP components have homologues in yeast as RAD genes.
These complexes for first the time were identified in patients suffering from Xeroderma pigmentosum (Xp). These proteins assemble in a sequence and locate the lesion in ATP dependent manner and cleave at both sides of the lesion and release 29 ntds long oligomers. Then PCNA, RF-C joins in ATP dependent manner, and then DNA pol-delta joins the complex for repairing the gap.
· In human systems there are seven complementary gene products named as gene A to gene G. They are involved in repairing UV induced dimer damage, bulk lesions and also inter strand links.
Human diseases caused by the above gene defects are-
· Xeroderma pigmentosum (XP), Cockayne’s syndrome (CS),
Trichothiodystrophy (CTTD).
Persons with XP disease are sensitive to UV, which causes blisters and ultimately cancer. Human excision repair system is called ERCC (excision repair components).
· Nucleotide excision involves, first the binding of XP-A to dimer of XP-F and ERCC. The XP-A recognizes and binds to the lesions. To such a site a replication protein HSSB binds. A general TF II consisting of 8 different proteins called TF-H, this complex of TFII-H is different from that of the TFII-H involved in transcription of structural genes. Two of the 8 protein complexes are XP-XP-B and XP-D. They are involved in excision, but also in opening of the ds DNA into a bubble during transcriptional initiation.
The entire complex is recruited to the lesion site by XP-A, where the complex is joined by XP-G.
· XP-G is an endonuclease. Among the endonucleases XP-F and XP-G, the XP-G makes a nick at 3’ end of the lesion and XP-F with ERCC-1 makes nick at 5’end about 25 ntds away from the lesion.
The TF-H as a complex perhaps acts as an unwinding complex and activates nicking process. The PCNA RF-C and DNA pol-delta join and release excinucleases subunits. This repair system can also remove inter strand linkages.
· In yeasts UV sensitive mutants have been recognized and the same have been isolated and they are called RAD group of genes.
RAD-3 group: involved in excision repair process, it also has helicase activity. It is also involved in post replication repair.
RAD-6 group: Involved in post replication repair process.
RAD-52 group: Involved in recombination repair process. There are two groups of RAD52 genes.
Group I: RAD 50, 51, 54, 55 and 57-homologous recombination repair.
Group II: RAD 50, MEE11 and XRS2- nonhomologous recombination repair.
In yeasts transcriptionally active genes are preferably repaired.
RAD 30: codes for DNA pol h
DNA polymerases involved in such repairs are DNA pol-IV and DNA pol-V coded for by din B and umuCD respectively
In yeast, most of the transcription repair mechanisms have been identified. The repair activity is concerned with actively transcribing genes and the damage in transcriptional strand is repaired immediately.
Removal of bulky adducts in new strands by Uvr-A, B, and C,
D mechanism:
Bulky lesions like Benzoyl (a) pyrene or Dialkyl Anthracene, Afflotoxin-B, 2-Acetyl aminofluorane, Methyl cholanthrene, UV activated Thymine dimers and many other chemo therapic drugs bind to DNA and cause distortions. Such DNA damages are removed by UVR-ABCD mechanism.
· Dinococcus radiourans produces UVR endonucleases, it is for this reason these microbes are highly resistant to UV damages.
Yeast also contains genes for repairing radiation damages called RAD genes.
· The UVR-ABC genes code for independent enzymes and assemble as complexes in a hierarchical sequence. Unlike photo reactivation photolyase enzymes, first UVR-A as a dimer and UVR-B as a dimer (?), form a complex and climb on to DNA and scan and locate lesions. Once they locate the lesion, they tightly bind and unwind DNA in the lesion region (ATP is required). Then the UVR-B, in this complex makes a cut or nick at 3 or 4 ntds away from the 3’ end of the lesion. Then UVR-C joins the complex and cuts at 5’ end of the lesion 12-13 ntds away. At this point UVR-D a helicase unwinds the cut strand and removes it. The DNA pol-I uses the 3’OH end of the nicked strand and fills the gap by polymerase activity. Finally the ends are sealed by ligase activity.
Repair of alkylated bases in the newly formed DNA strands:
There are many different kinds of alkylating agents, which can methylate guanine at 6’O –position and the 8’O of –guanine; such guanines can easily base pair with Adenine and cause missense codons or it can create a nonsense codons. Cytosine can be methylated or adenine can be methylated.
· A specific enzyme named as O-6 methyl guanine DNA methyl transferase removes this bulky methyl group. This enzyme is produced by Ada group of genes. These genes produce not only methyl transferases as well as glycosylases. This Ada-protein consists of 354 amino acids. This enzyme can perform two related functions. The C- terminal region can remove methyl group from Guanine, and transfer it on to its own enzyme surface at cysteine residue at 321 aa position (the cysteine is located at carboxyl end). The N- terminal region of the protein removes methyl group from phosphate of CH3p-s-CH3p- backbone, however the methyl group from phosphate is removed onto its own cysteine at 69th cysteine amino acid. While doing so the enzyme flips the base out of helix, on reapir it is flipped back to its position.
These 'Ado' related enzymes are inducible enzymes. When cells are grown on lower doses of nitrosoguanidine or such compounds, cells become resistant to alkylating drugs, this is because, cells produce both the enzymes at increased concentration. Once the enzymes transfer methyl groups on to their surfaces, they are not recycled, but they are degraded and fresh enzymes are provided for the functions.
Recombination repair system:
In most of the repair mechanisms, discussed above, the damage is in one of the two strands. Such damages can be found in the newly made strand or it can also be found in parental strands. Such damages do not mean that the information is completely lost, because the correct information is found in one of its complementary strands. Lesions may be found in both the strands, but at different locations. Here the repair process involves recombination and post-replicative repair.
List of genes involved in recombination repair
|
Recombination events |
Prokaryotes |
Eukaryotes |
|
Pairing of homolog- ous strands, strand invasion |
RecA |
RAD51, DCM1 |
|
Induction of DSB |
None |
Spo11 in meiosis, HO –mating switch |
|
Processing DNA ends to generate ss strands |
RecBCD-helicase/ nucleases |
MRE11,XRS2 proteins RAD 50/51/54/ 57/58/60. |
|
Assembly of strands Exchange proteins |
RecBCD &RecFOR |
RAD 52, 59 |
|
Holliday junction Recognition and Branch migration |
RuvAB complex |
Not known |
|
Resolve of Holliday Junction |
Ruv C |
Perhaps Mus81 And others |
Pol B (dinA)- encodes polymerization subunits of DNA pol-II.
Uvr A and Uvr B- encodes uvrABC Excinuclease subunits.
Umu C &D – encode for DNA pol-V D2C complex (umu-stands for unmutable).
Sul A- inhibits cell division and allows time for repair.
Rec A- it is required for error prone recombination repair.
Din B-encodes DNA pol-IV.
Him A-subunit of IHF, involved in site specific recombination, replication, transposition and gene expression. The Integration host factor is found in bacterial systems as well as eukaryotic cells. When it binds to DNA, it makes the DNA bend by several degrees, this will help the factors bound to DNA at different position to come close to one another and facilitate interaction.
Rec N- recombination repair (exact functions not known).
Din D and din F- functions not clear.
E.coli homologous recombination repair requires:
Proteins Involved:
Rec-B, rec-C and rec-D- they have both helicase and nuclease activities; they bind to the free ends of DNA (broken), move inwards and degrade ends in ATP dependent manner. When the complex reaches 5’ end of the Chi sequence- CTGGTGG, it changes its activities, it reduces 3’ end degradation and enhances degradation from 5’ end, thus creates 3’ overhangs. E.coli contain not less than 900 such chi sequences.
Rec A promotes homologous recombination by pairing to dsDNA, forces dsDNA to open and this strand intrudes and searches for homologous regions and helps in branch migration in 5’>3’ direction and facilitates in forming Holliday intermediates. RecA forms RecA filament by cooperative binding to single stranded of DNA (thousands of monomeric subunits bind), and generates coiled structure with six monomers per turn. This filament pairs with ds helix, intrudes into the helix and unwinds and searches for homology. Rec F, Rec O and Rec R regulate the assembly RecA filament
Ruv A and Ruv B another set of proteins bind to Holliday junctions, displace RecA and promote branch migration.
Ruv C- is nuclease that cleaves and resolves Holliday junctions that are developed after recombination events.
Such deleterious and harmful defects are also repaired by UvrABC proteins by what is called recombination repair.
Here the repair is more or less perfect, and there is no room for any mistake. These repairs are made at post replication stages or repairs are made when cells are in resting stage, where cells are not dividing.
Any damage to DNA in the form of break or dimers or heavy adduct in a replication DNA induce SOS responses, where Ruv A and Ruv B are synthesized in in greater amounts.
First possible method:
If there a break in one of the strands, during replication DNA pols halts well before they encounter the site of damage, dissociate and reassociate farther away(?). Thus a gap is left behind in one of the replicating strands.
If the DNA pol encounters such dimers they can skip the damaged site by dissociation from the replicating strand. But the replication of the other strand contnues normally, (how?). In such situation homologous strand against the gap region is used to repair the broken strand or the strand with lesion.
In this process instead of the replication fork collapsing, it regresses to form a Holiday junction (chicken foot like). This process is mediated by RecA or Rec G. The Rec G is a ATP dependent helicase and it catalyzes branch migration at DNA junctions. Reverse branch migration is performed by RuvAB or RevG, which in fact generates reconstituted replication fork and replication continues with out repairing the lesion. Later the lesion is removed.
The second possible method:
In this a single stranded nick when encountered Replication stops and DNA pol III dissociates from the form only from the strand that is nicked or gapped. Its repair is performed by RecBCD and RuvA and RuvB. Rec A uses single strands and coats it with its monomers. Such RecA coated strands invade the homologous dsDNA. The gene products involved are RecA, recBCD and Rec-F. RecA forms a filament with coated monomers, it leads to strand exchange and recombination events lead to correct the damaged strand. Rec BC is involved in reinitiation replication fork. RecF is involved in repairing the gap. Rec-F consists of Rec-O, rec-R; they form RecOR and RecOF pairs and repair is performed by them. Branch migration is aided by Ruv AB, that produces a Holliday junction. RuvC resolves the Holliday junction.
Repair of double stranded DNA Break:
Double stranded breaks are repaired by non-homologous recombination and homologous recombination methods.
Homologous recombination repair mechanism:
In yeast systems DSB is repaired by gene products such as Rad50, Mre11 and Xrs2 and few others. First they create 3’ single stranded DNA. Then gene products such as Rad 51, Rad 52, Rad 54, Rad 55, Rad 57 and Rpa1 are involved in strand invasion into homologous region of the normal ds DNA For this kind of repair it requires homologous DNA which is provided by conjugation. This leads to synthesis and branch migration. Such recombination methods do produce Holliday junctions. They are then resolved by resolvase and a complex of proteins such as Msh2, Msh3,Msh6,mlh1 and Pms1. in this repair the information is not lost and repair is error free.
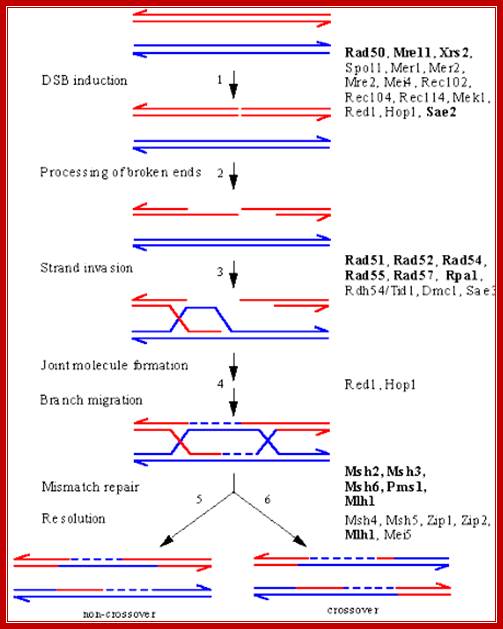
Meoiotic DSB Repair in Yeast:
An overview of meiotic recombination between two homologous chromosomes, based on the DSB repair model, showing intermediates and the proteins implicated in their formation by genetic and/or molecular criteria. The points at which several proteins act is still under investigation, and some may be required at several steps. Proteins that function in both mitotic and meiotic HR are indicated in bold; all others are unique to meiosis. A DSB is introduced in a DNA duplex by the Spo11 nuclease, likely acting in conjunction with several other proteins that are known to be required for DSB induction. The 5' ends of the break undergo 5' to 3' resection to yield 3'-OH single-stranded tails. 3) One of these single-stranded tails invades a homologous duplex, displacing a D-loop. 4) Several steps resulting in the formation of a bimolecular intermediate with double Holliday junctions follow. At this point, correction of mismatches in heteroduplex DNA can result in gene conversion. 5) Resolution of the intermediate to yield a product with a parental configuration of flanking sequences (non-crossover). 6) Resolution to yield a crossover product..The above diagram illustrates ds break recombination repair in Yeast systems, and note the gene products involved in the repair process. http://atlasgeneticsoncology.org/ http://atlasgeneticsoncology.org
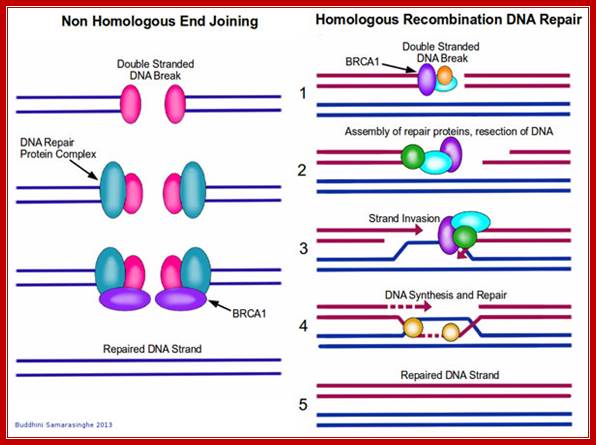
Nonhomologous exchange recombination repair (NHER):
Broken ends are recognized and bound by a set of proteins such as Ku70 and Ku78 (80) as heterodimers and act as scaffold. They hold the ends together. Then they are joined by protein kinases and some exonucleases to produce jagged ends or blunt ends. The Ku helicases unwind the ends into short single stranded regions, which provide base pairing between the jagged ends and use micro-homology. Unpaired 5’ ends are removed and ligase seals the nicks. In this process the recombination is not homologous and perfect. However sealed DNA ends do not evoke responses that have far reaching consequences to the cell.
http://blogs.scientificamerican.com/

The diagram shows the realignment of the ends, microhomology with which they base pair and ligate thus the nonhomologous ends are fused. https://www.circuitwiringdiagram.info
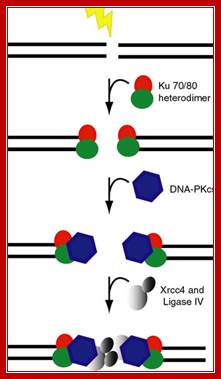
In higher mammalian systems the repair of double strand break is performed by a couple of proteins called Ku70 and Ku 80, which act as scaffold and hold the ends of the broken DNA, then recruit proteins required for repairing the broken end either by exonuclease activity or by end filling to generate blunt ends. Then the special ligase ligates the ends, this may have some changes in the sequences between the joining ends, but this kind of somatic recombination is in vogue with respect to immunoglobulin DNA whicle they perform somatic recombination of DNA. leanfytiini-kirjallisuutta.blogspot.com
Such damages involving both strands are repaired by a mechanism called recombination repair. Among several recombination repair mechanisms, Interstrand excision repair, Translesion recombination and Error prone repair mechanisms are significant.
DNA Repair at Transcription stage:
During transcription transcriptional enzyme complex may come across lesions such as T-T dimers, heavy adducts or modified bases in the template strand. Such damages have been left without repair or escaped from repair process.
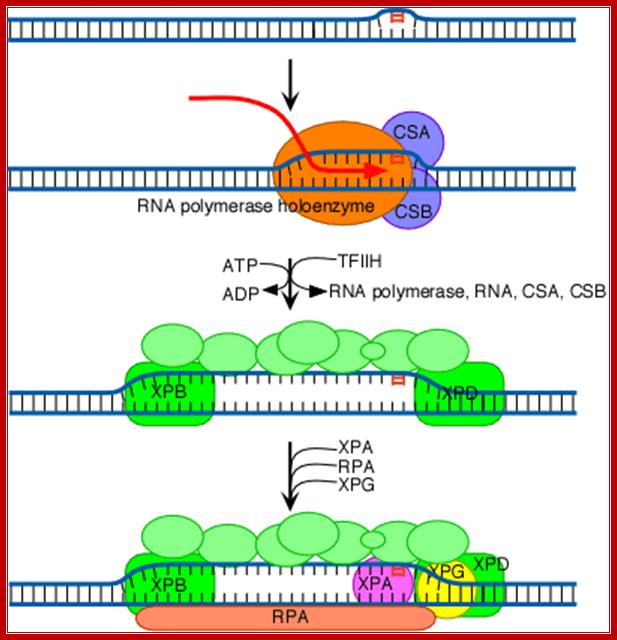
The increased distortion produced by XPC/HR23B permits the entry and binding of the general transcription factor TFIIH, whose 10 subunits are colored in various shades of green in the above diagram (see also the table above). Two of these subunits (XPB and XPD; shown in brighter green) are helicases, which bind to the damaged strand and use the energy of ATP to unwind a stretch of 20-30 nucleotides including the damaged site.
The above diagram shows the involvement of multiprotein complex in repairing the damaged template strand during transcription; TFII H components play a significant role in this process. http://joelhuberman.net/
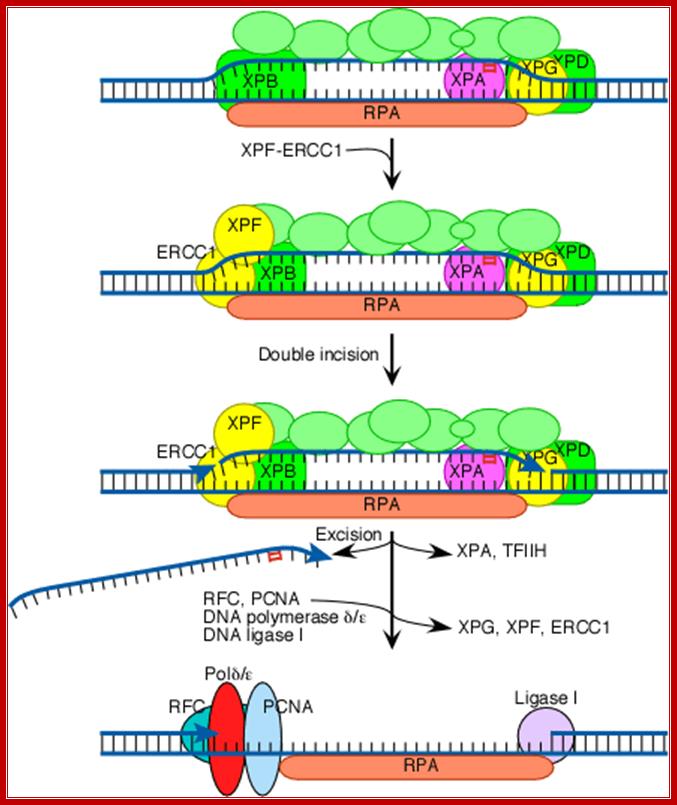
Both XPG and XPF-ERCC1 are specific for junctions between single- and double-stranded DNA. XPG, which is closely related to the FEN-1 nuclease that participates in base excision repair, cuts on the 3' side of such a junction, while ERCC1/XPF (a heterodimeric protein complex) cuts on the 5' side. The cut made by XPG is 2-8 nucleotides from the lesion, and the cut made by ERCC1/XPF is 15-24 nucleotides away. The cuts are paired with each other (probably as a consequence of the structure of the multiprotein complex) in such a way that the damage-containing oligonucleotide between the cuts averages 27 nucleotides (range 24-32 nucleotides). Next the replicative gap-repair proteins, RFC, PCNA, and DNA polymerase delta or epsilon, bind to the 3'-OH group (arrowhead) generated by the ERCC1-XPF cut, and they carry out new DNA synthesis that fills the gap. This leads to displacement of the damage-containing oligonucleotide and of TFIIH, XPA, XPG, and XPF-ERCC1. The final nick is sealed by DNA ligase I.http://joelhuberman.net/
Such situations are rare but transcriptional apparatus may encounter such blocks. In yeast cells DNA is repaired preferentially and remove impediments for smooth transcription. In yeast cells the genes involved in repair of the DNA at the time of transcription are RAD genes. They are the homologues of mammalian systems. In fact RAD3 is always associated with RNApol complex.
In E.coli a protein by name Mfd is involved in excision repair of the template strand. Mfd joins RNAP, which has been stalled at the site of lesion and displaces the RNAP and recruits Uvr ABC to the damaged site and repair is executed. Once the repair is over another RNAp assemble and transcription reinitiates from the start.
In mammals following UV induced damage an alternate form of TFII-H recognizes the lesion and creates transcription bubble, the TFII-H is a helicase form not the other TFII-H that is involved in initiation transcription. The H complex consists of a core complex made up of 5 subunits, plus it has some more components involved in repair process, a total of amounting to 8-10 subunits. Among them are XP components. They are XPC(repair enzymes), endonucleases such as XPD, XPG, XPF,ERCC1; all in all there 7-8 XP subunits named as XP-A to XP-G. Another subunit associated with this complex is XP-V, it codes for DNApol-h, which is responsible for the removal T-T dimers; skin cancer is mainly due to mutation in XP-V gene. Its homologue in yeast is RAD3-D.
In humans they are called XP for mutation in these cause a disease called Xeroderma pigmentosum, a deadly disease can cause skin cancer and highly sensitive to sun light. In the repair process the large subunit of RNAP gets degraded (how?) as it dissociates from the template.
The repair form of TFII-H locates the lesion and the core complex binds and other subunits join the core complex. Then H-complex creates a transcription bubble. To begin with XPD and XPB as heterodimers bind to the lesion. XPD provides helicase activity and give stability to the H-complex. It is this complex, perse, opens the DNA into a bubble, call it a Repair bubble. This bubble is now joined by XPG and binds to DNA. It cleaves the templates strand towards 3’ side of the lesion. It is at this juncture ERCC1 joins the complex and it cleaves at 5’ side of the lesion and removes a reasonably chunk of template strand. Excision of the damaged strand makes the H complex to dissociate from the damaged site. Then DNA pol-d-e joins the site assisted by RFC, PCNA and DNA ligase1. The new strand is synthesized and ligase seals the ends.
Mammals and Human genes:
In mammalian systems including humans, following UV damage, TFII H and another complex of TFII H (one of the XP) creates a transcriptional bubble. Otherwise the TFII-H core consists of six subunits and it has a role in elongation of the transcriptional initiation by using its kinase property.
TFIIH consists of core complex. This is common for the TFIIH for both situations i.e. for normal situation and DNA damaged situation.
The TFII-H complex of the first type is involved in elongation of the first 50 or so nucleotides after initiation of the transcript. This is achieved by its kinase activity.
The TFII-H complex in DNA damaged conditions, when encounters the damage, it is replaced by the second core complex with its associated factors for the repair functions.
When the transcriptional complex encounters the damage site, the large subunit of the RNAP complex gets dissociated and degraded (?)
The TFIIH complex involved in DNA damage repair consists of core complex and other XPC repair enzyme components. The later include endonucleases, XPG, XPF, ERcc1.
In yeast systems they are named as RAD members.
In humans, mutants of the above kind are called XP. Mutation in XP cause Xeroderma pigmentosa, which in course of time can lead to skin cancer called Cockayne’s disease (refers to DNA repair disorder). This is due to mutations in CSA and CSB genes, which are the part of TFII-H complex. This disease is also caused by mutation in XPD, which is a pleotropic protein, but is required for the stability of TFII-H complex involved in DNA repair of the transcriptional template at the time of transcription. It provides helicase activity at the time of repair.
In the case of Damaged DNA, the XPD and XPB of TFII-H complex binds to DNA scans for the damage and binds. This complex with its helicase components opens the DNA at damaged site into a bubble, which contains the damaged site.
Then XPG binds this complex of TFII-H and displaces it, and cleaves the damaged strand at its 3’ ends of the bubble.
Then ERcc1 assembles the complex and cleaves at the 5’ end of the bubble. The cleaved patch is removed and repaired by new synthesis of the gapped strand.
XP mutation in human cultured fibroblast cells fails to repair UV damage and repair other bulky adducts.
XPA to XPG 7-8 genes involved in DNA damage repair.
Human XP –V is equivalent ot Yeast Rad3D. XP-V is DNA pol-h, which is responsible for removal of T-T dimers
Xeroderma pigmentosa defect is autosomal. There are seven complementation groups. Clinically they are found to be sun sensitive, show dermatoses, hyper pigmentation, retinal degeneration, neurological disorder, mental disorder and retardness,, can lead to cancer. All this is due to malfunctions or non functioning genes involved in damaged DNA repair.