DNA: DNA and DNA: RNA Hybridization Kinetics:
Reassociation Methods;
·
Use a DNA which is fragmented to a size of 3x10^2, (5x10^2 ?) bp long.
The concentration should be optimal for reassociation (should be calculated in Molar values). Each of the genome fragmented sequences should be more than thousand copies or more. One can calculate the number of copies by knowing the genome length and the concentration one has taken. Calculate OD into micrograms, micrograms into picograms. 1pg = 978 x 10 ^6 bp or 0.978x10^9 bp.
The DNA isolated should be pure free from RNA and proteins, and the same should be suspended in 0.15M sodium phosphate buffer pH6.8 (today you have different composite buffers).
The reaction and its rate depends upon the number of each segment and frequency of each segment with which the complementary strands react and reassociate; and the buffer factor also contributes to the rate of reaction.
The sheered DNA (~400 to ~500bp long or more) is heated to 85⁰C (melting temperature is called Tm, it varies from one genome to the other) till it melts into ssDNA (denaturation) and immediately cool it to prevent reannealing.
· Such ssDNA in a solution is allowed to anneal or complementary strands to reassociate with each other in a suitable buffer (0.15M Na Phosphate buffer) at a temperature 25⁰C less than its melting Tm (Tm is the temperature at which ds DNA melts into single strands). Complementary strands reassociation is by random collision, when a strand collides with another with a strand having right complementary sequences they start base pairing. This process takes time, really good time.
Take OD at 260nm to measure reassociation at a determined time intervals. Or the hybridization can also be quantified using hydroxyapatite columns (HAP) or nitrocellulose membranes or glass micro tubes with nitrocellulose where ds DNA retained in bound form and single stranded DNA remains unbound and comes out of the column or the membrane, and hybridized DNA can be eluted and measured quantitatively.
So the reassociation reaction is measured at certain time intervals by pouring the solution into the hydroxyapatite column and allowed dsDNA to bind. Then the column is washed to remove unbound ssDNA. Then the bound dsDNA is eluted and quantified.

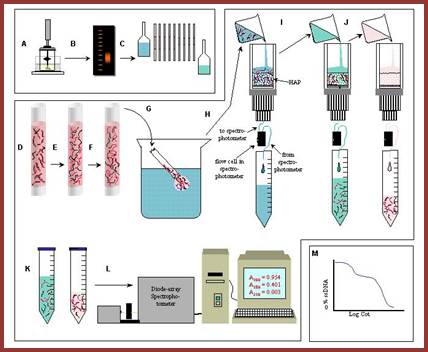
DNA microarray co-hybridization experiment; Automation is used; Yevgeniy Grigoryev; Top Fig; http://bitesizebio.com/;bottom fig.
Refer to; overview of cot analysis; http://plantgenome.agtec.uga.edu/
The percentage of dsDNA reassociated is quantitated against original concentration (Co) and start time called zero (t^o) and the same is plotted as the function of the amount of fragments reacted at given time elapsed. This is a measure of ‘product of ‘Concentration’ (quantity) and ‘Time’ (S^1L^1), which is referred to as ‘Cot’ value.
A similar reaction is performed with a DNA whose size and sequence is known, ex. E.coli DNA, whose size 4.6x10⁶ bp, and sequences are known. Any such DNA can be used as reference DNA or standard DNA whose Cot value has already been determined. Most of the E.coli DNA sequences are unique and non-repetitive.
Graphic representation of reactions gives a pattern of curves called Cot curves, from which one can obtain Cot ½ values where the concentration of hybrids formed at half the time during reassociation. If the complete hybridization takes 72hrs or 144hrs measure at the interval of 36 hrs and 72hrs that gives ¼ and ½ times. Concentration of dsDNA at half time gives Cot ½ values.
The rate of reaction depends on length and the complementarity of the reacting fragments and the copy numbers of each fragment or sequence, for example, take three different kinds of DNA fragments.
· First one consists of unique sequences, differ from each other, each of the sequence have a length ranging from 500 ntds to 1000 ntds or more. Each of these sequences has copy number of one or two.
The second class of fragments contains sequences of 300 or more bp and they are repeated 100 to 200 or more times.
· The third class of DNA fractions consist of short sequence, two to 50ntds or little more but repeated 500 to thousands times.
Complementary strands with high copy numbers collide with higher frequency and reassociate faster, but complementary copies with low copy numbers base pair slowly for the frequency of collision between the right complementary strands is lower. Thus kinetics of reassociation behaves as if each of them is different but specific reaction orders.
· The first class of DNA fragments, on the other hand, where the sequences are unique and they are found only as one or two copies each, the reaction rate is very slow for it is difficult to find the unique complementary sequences with ease and facility; so base pairing takes more time than the others.
The second fraction or second class of DNA that has a moderate copy numbers, which reacts or reassociate at moderate rate and falls in between the first and third.
· The third DNA fragments are shorter, similar sequences, each sequence consist of large number of copies, hence the reassociation reaction is faster because of high copy number, the frequency collision between similar copies is very high and reassociation reaction is fast.
The Kinetic order of reactions is classified into First order, Second order and Third order. Further subdivisions are Psuedorder and Zero order. Kinetic reactions have thermodynamic properties.

Genetic similarities: Wilson et al; Diagram shows how the denatured single stranded DNA strands reassociate using sequence specificity. If the hybridizing DNA sequences are very closely related one finds few mis-pairing; http://evolution.berkeley.edu/
Equations used to determine Cot values:
D/Do = -K . Cot,
Cot1/2 = 1/K
‘Do’ concentration at zero time, D concentration at specific time of reassociation. K is rate constant, and Cot is the product of ds DNA concentration (in molar values) and time taken for such reaction. Cot1/2 is the value at half time.
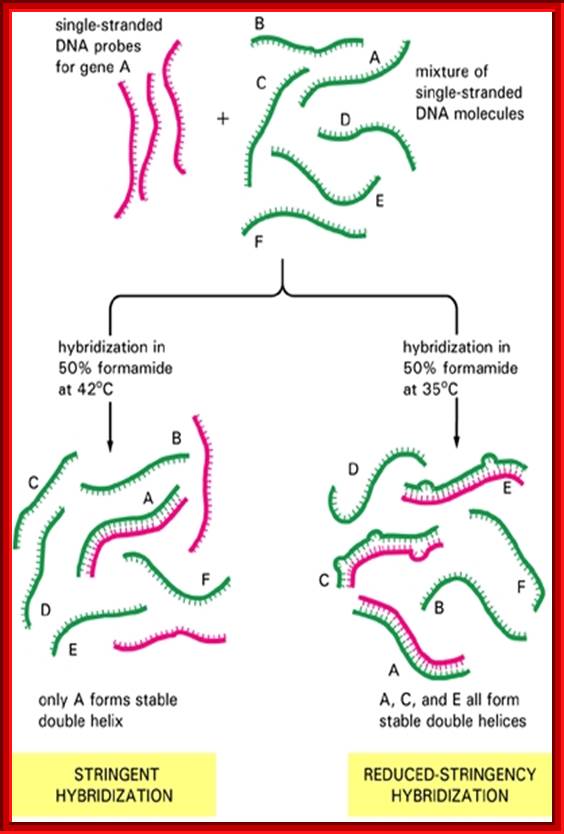
Recombinant DNA tech and Med: Specificity of hybridization is controlled by temperature; http://quizlet.com/
Kinetic-Rate Orders
|
|
Zero-Order |
First-Order |
Second-Order |
nth-Order |
|
|
|
|
|
|
1. Zero-Order Reactions: Zero-order reactions (order = 0) have a constant rate. This rate is independent of the concentration of the reactants. The rate law is: rate = k, with k having the units of M/sec. Examples- enzyme reaction; the reaction is independent of concentration of the substrates.
2. First-Order Reactions: A first order reaction (order = 1) has a rate proportional to the concentration of one of the reactants. A common example of a first-order reaction is the phenomenon of radioactive decay. The rate law is: rate = k[A] (or B instead of A), with k having the units of sec.
3. Second-Order Reactions: A second-order reaction (order = 2) has a rate proportional to the concentration of the square of the product of the concentration of two reactants.
4. Mixed-Order or Higher-Order Reactions: Mixed-order reactions have a fractional order for their rate; rate = k[A]^ 1/3
First order kinetic equation:
Reactions involve only one substrate.
Dx / DT = k (a – x),
Where ‘a’ = concentration of ‘A’ molecules,
X = concentration of molecules that have undergone reactions to products, example radioactive materials undergo decay by their atomic instability.
K = 1 /t ln a /a-x,
Rate constant: K = 2.303 / t log [A] 0 / [A] t,
K is the rate constant,
K dt = dx / a-x
Or it can be written as D /Do = - K C0t
Acronym Do is an initial concentration at zero time, D is the concentration after a time period of reaction, K is the rate constant.
These kinetic reactions can be used in DNA driven or RNA driven reaction mode, where one of the component is far excess, which can be considered as pseudo first order kinetics. This can be rewritten as-
D / D 0 = C0t1/2 = 0.5 or K = ln2 / K x t ½,
If the reaction is driven by RNA it can be written as R0t1/2 = ln2 / K, where Ln is natural log of 2.
Second order kinetics:
Reactions involve two reactants of similar kind or Different kinds.
dx / dt = k (a- x )^2,
dx / (a - x )^2 = k dt,
Integrated equations gives-
K = 1 / t. x / a (a-x),
K = 1 / conc. x 1 / time
K = 1 / mol. liter x 1 / time
K = Mol L^-1 time S ^-1,
Mixed or Third Order Kinetics:
In this, more than two reactants take part in the reaction, which is really rare but not uncommon.
dA + dB + dC = XYZ (?)
rate =
k[A]1/3 ;![]()
Pseudo-order kinetics:
In a bimolecular reaction one of the reactants is far excess in concentration and its concentration remains so. For example a small amount of Na Cl is added to one liter of water, where water is excess, in such cases
The rate = k[A]1/3
The rate = K [A][B]
Rate = K’ [A]
New rate constant K’ = K [B].
Actually this is a second order but in practical terms, because of excess of water it considered as pseudo first order. Ex. Hydrolysis of an ester or sucrose in water.
Zero order:
The rate of reaction is independent of the kind and concentration of substrates or reactants. Rate concentration is equal to the rate of reactions at all concentrations.
dx / dt = - d ( a – x ) / dt.
K = d [A] / dt = mol / dt x 1 / time
Example: In enzymatic reactions where the substrate concentration is far excess, but the rate of reaction independent of substrate concentrations.
DNA-DNA Reaction Kinetics:
The rate of reassociation of complementary DNA strands is determined by the kinetic equation, which is of second order.
dC / dt = - K C^2,
C is the concentration of ss or ds DNA at a time t,
Where dC means the difference (delta) in the concentration of ss or ds DNA at a given time (dt) DT. And K is the rate constant.
The equation can be rewritten to determine the rate of reassociation after specific time intervals.
The equation is C / C 0 = 1 / 1 x K.Cot ½,
The Cot 1 / 2 indicate the reassociation is half complete at a time called t1/2.
By measuring ss or ds DNA at zero time i.e. Co, and time t° and the concentration of ssDNA at another time t after a period of reaction; the kinetic equation can be written as-
C / Co = 1 / 1 + K. Cot,
Where C is the concentration after a lapse of time t,
The product of Co (conc. of ssDNA at zero time) and the time t (period of incubation) is,
Co x t = Co t.
When the reaction is half complete it is written as –
C / Co = 1 / 2 = 1 / 1 + K. Cot 1 / 2,
So the Cot 1/2 is equal to 1/ K. Hence the Cot ½ is the product of conc. of ss or ds DNA left after reassociation into ds forms and the time required for reassociation to half completion. It can be rewritten as, the Cot ½ is the product of conc. of dsDNA after reassociation and the time required for reassociation to half completion. Cot ½ implies the rate of reassociation, which means faster the reassociation smaller the Cot ½, and the slower the reassociation larger is the cot ½ values. This indicates, segments of DNA with a particular sequence, if contain hundreds of thousands of copies, the frequency of collision is greater, and they reassociate at higher frequency (smaller Cot1/2) and those fragments which are longer and have one or two copies with unique sequences reassociate slower, sometime the reaction does not complete, and one has to use computer programming for getting the data.
Reassociation reaction data can be graphically expressed as Cot curves, by plotting a fraction of DNA reassociated (1 – C / Co) against the log of Cot. The rate constant is expressed in moles of nucleotides per liter per second; whereas the Cot ½ is expressed as nucleotides moles x sec / liter. The rate of reassociation is inversely proportional to the length of the re-associating DNA.
Re-association reactions of any DNA from any source provide information about different reacting components of the said genome, irrespective of their sizes, which tells us which component contains what, and what component performs which function and this perhaps explains the C-value paradox to some extent.
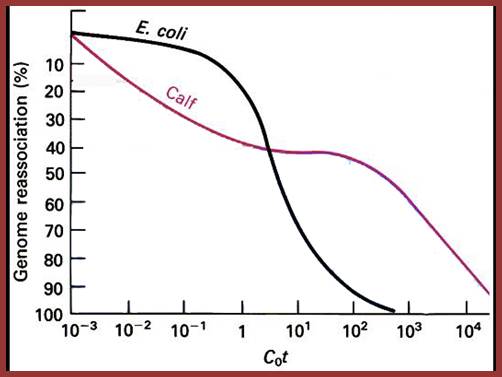
Reassociation kinetic curves expressed as cot values expressed as graphical representation in the form of Cot curves. This figure shows reassociation cot values of E.coli and calf thymus D DNA, where DNA was sheared into 1000bp long fragments and denatured by heating into single strands, then they are allowed to reassociate according to their complementary base pairs at stringent conditions. The extent of reassociation i.e. ₁percentage of the total was measured by chromatographic techniques and plotted against Cot i.e. molar concentration x time in seconds. Calf thymus DNA reassociate as different fragments, some fast, some median and some slow, but E.coli DNA behaves as the last mention composition; slow component; Kenneth G. Wilson,http://www.cas.miamioh.edu/
Meanings of terms:
Chemical complexity: The size of a genome is called chemical complexity, ex: E.coli- 4.2 to 4.6 x 10^6 bp or this can be expressed in pg i.e. 0.0045 pg for E.coli genome. T4 Phage- 1.7 x 10^5 bp = 0.00016 pg; 1pg-1x10^12 of the gram.
1 pg of ds DNA = 6x10^11 daltons = 0.978x10^9bp (old) or 978X10^6bp (new); 1pg = 0.978 x 10^9bp
Kinetic complexity: It gives the value for the total length of each reassociated DNA fragments. This is referred to as complexity expressed in base pairs. It can be obtained by dividing Cot1/2 of the unknown genome by the Cot1/2 of any standard or known DNA such as E.coli DNA. Or complexity of any unknown genome divided by complexity of the known genome value.
Cot ½ of a reaction, therefore suggests that the total length of different sequences present in the gnomic DNA. Renaturation of any genome displays a Cot1/2 that is proportional to its complexity (length of different sequences). The complexity of any genome can be determined by comparing the Cot1/2 of it with that of Cot1/2 of known genome; here we use E.coli genome as the standard DNA.
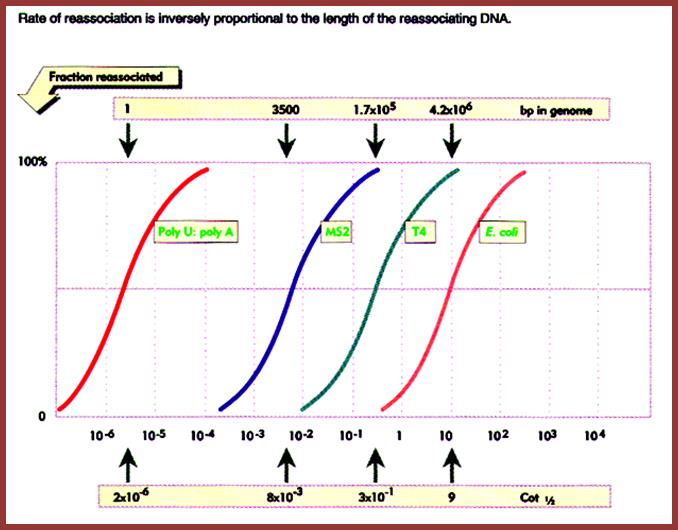
The diagram shows the rate of reassociation is inversely proportional to the length of the reassociating DNA of different systems such as Pol-U - Pol A oligomers, MS2 genomic DNA, T4 phage DNA and E.coli DNA. This graphic depicts the size of the genome and the rate with which they reassociate is expressed in the form of kinetic components and represented as Cot values, from which Cot1/2 values can be determined. Mol.Biol. http://biosiva.50webs.org/rep1.htm
Cot1/2 of As with Us = 2.1x10^- 6, Chemical complexity =1 bp.
Cot1/2 of MS2 phage = 8x10^-3, Chemical complexity = 3500 ntds.
Cot ½ of T4 phage = 3x10^-1, Chemical complexity = 1.7x10^5 bp.
Cot1/2 of E.coli = 9, it is all non-repetitive; Chemical complexity = 4.6x10^6, this amounts to 45% of the non-repetitive DNA; which means Cot1/2 of E.coli DNA i.e. - 9x0.45 = 4.05.
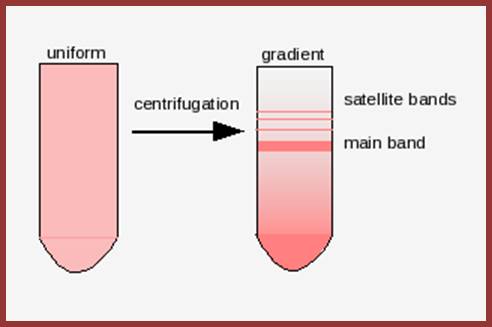
Reassociation kinetics http://home.cc.umanitoba.ca/
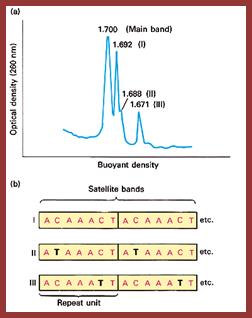

Satellite bands appear on ultracentrifugation as distinct bands due to their repetitive tandem sequence segments of specific DNA sequences. They appear as separate smaller peeks, different from the main peak; hence they are called satelliteDNA.;
J.G. Gall and D.D. Atherton, J. Mol. Biol. 85; http://home.cc.umanitoba.ca/
Genomic DNA Analysis- Charts:
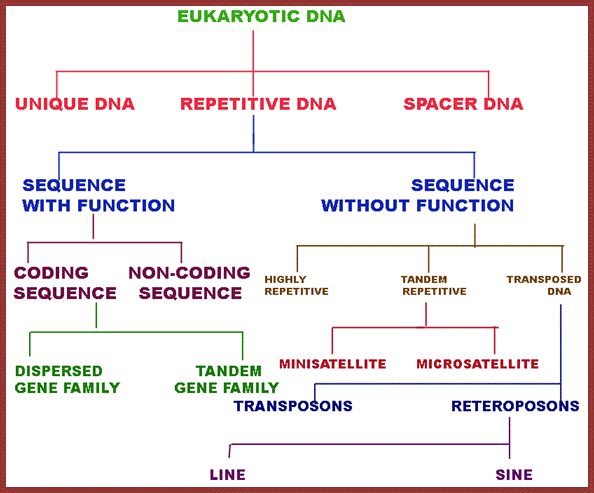
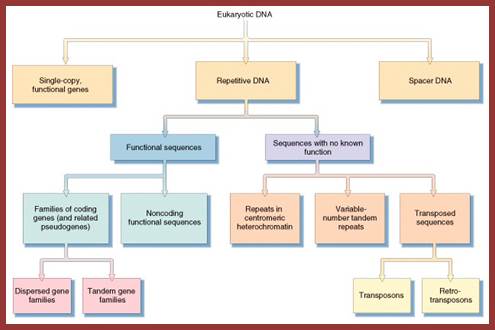
This data in the form of flow chart shows the kinds and class of DNA. http://biosiva.50webs.org/
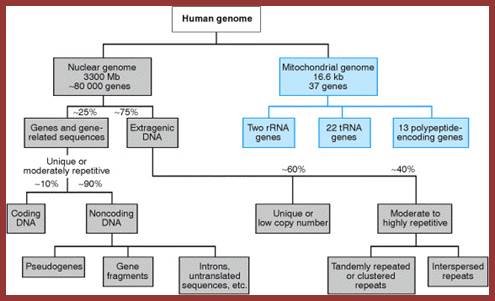
http://www.mun.ca/
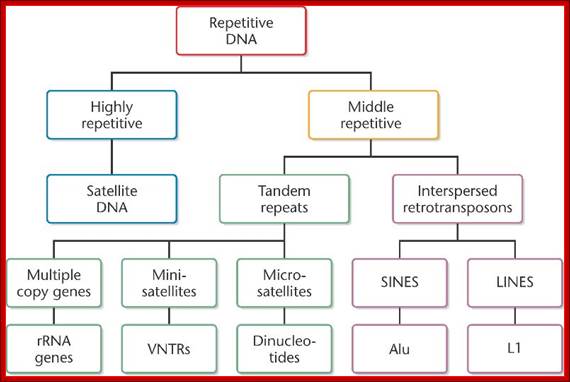
The above flow charts show various components of repetitive DNA. The top colored chart is not visible so the black and white chart is given; here; repetitive DNA can be grouped into many categories. http://bio3400.nicerweb.com
Highly repetitive DNA: Certain sequences, generally short sequences of 2-4, 10 to 100bp long, if they are repeated several thousand times, then its cot1/2 value is very low for they reassociate quickly.
Moderately repetitive DNA: This kind is not so highly repetitive DNA, where the DNA sequences are moderately repeated to about 200 to 300 or little more number of copies; its Cot1/2 value is moderate.
Nonrepetitive DNA: It is also called unique sequence DNA, which actually code for proteins and their copy numbers is just one each or two, maximally 4-5, its Cot1/2 value is very high.
Eukaryotic DNA consists of a variety of components and the same is given in drop down format one above and the other below.
Use a genome, for example, of a size 7x10^8 bp that is its chemical complexity. The genome is sheered into required small fragments and then they are subjected to reassociation reactions using standard buffers and temperature. Reassociation reactions are measured in terms of changes in ssDNA or dsDNA concentration with time lapse. Based on graphic representations, their cot1/2 is determined. Data for E.coli genome is available and it is used as the reference or standard Cot ½ values.

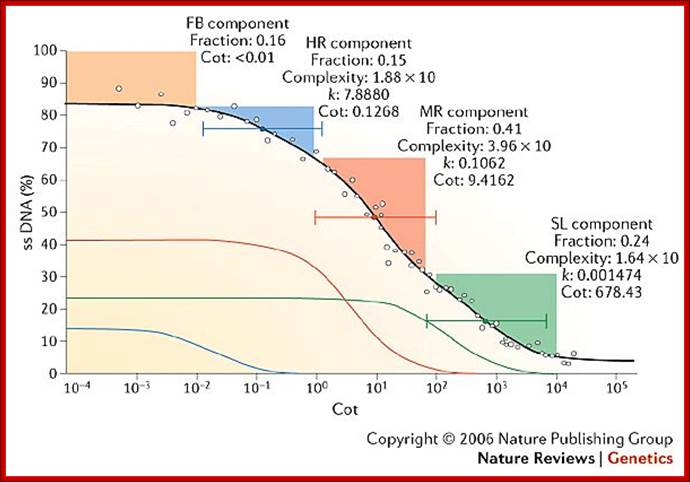
This reassociation cot curve and reassociation kinetics shows three different rates and complexities, and they look like independent kinetic components; such as Fast component, Intermediate and Slow component and each components represents a percentage of the genome that is used for the reassociation experiments. The chemical complexity of the DNA used in this experiment is 7x10^8bp;Y axis- fraction reassociated 1-C/Co; X axis- Cot values from it one can get Cot1/2 values; http://biosiva.50webs.org/rep1.htm
Observing the graphic pattern of Cot1/2 values shown above one can observe three distinct kinetic phases and each looks like independent components. The first, second and the third components are clearly separated from one another. Each kinetic component represents different proportion of the total genomic DNA that gets reassociated. In addition to these three components one can add another component which is independent of DNA concentration; it is called fold back (FB) DNA for it consists of palindromic sequences and fold back DNA base pair immediately.
Analysis of Kinetic Components:
How to determine Kinetic Complexity:
The Cot value of the E.coli is 9; here the DNA behaves like a slow or unique component. E.coli DNA is used, as the known or standard DNA, for its Cot1/2 value has been determined or for that matter any bacterial DNA cot value can be determined.
In the experiment 7.8x10^8bp DNA is used; the above graphic shows the slow component, represents 45% of the total genome. E.coli genome’s Cot value is 9 and 45% of the 9 is 4.05 (9.0 x 0.45 = 4.05). Similarly one can calculate for other components.
Kinetic complexity of each component can be determined first by multiplying the size of standard DNA, percentage of the component it occupies and the Cot1/2 of that component. Then it is divided by the Cot ½ of the standard DNA. The dividend of this is the kinetic complexity. Using the experimental data calculations are made.
First component: Size of the genome used for the experiment is 7x10^8 bp. The proportion of the reassociation as shown in the diagram is 0.25, so 0.25 X Cot1/2 of the said component (0.0013) divided by the Cot1/2 of the standard DNA which is derived from multiplying the Cot1/2 of the standard DNA (9.0 X 0.45 = 4.05) divided by the percentage it occupies as kinetic component is equals to 340bp.
This is the fast kinetic component with short sequences having high copy numbers. They are found as Satellite DNA, Telomeric DNA, CEN DNA and heterochromatic DNA, each of which don’t code for any proteins. Some may code of small Mol.wt noncoding RNAs. These sequences are interspersed all over the length of the chromosomal DNA. They are involved in hetero-chromatization and centromeric organization; some are involved in the attachment of chromosomes to proteins such as scaffold proteins (SARs) and some may attach to nuclear laminins too? This fraction contains highly repetitive sequences.
~4.6 x10^6 bp x 0.25 x cot1/2 x 0.0013/ 4.05 = 340
Second component: ~4.6 x 10^6 bp the size of the standard genome x 0.3 (percentage of the genome) x 1.9 Cot1/2 value, divided by 4.05 = 6.0 x 10^5 bp. This has reasonably moderately longer sequences, but has moderate number of copies each (moderately repetitive DNA). Ex: rRNA, tRNA, 7SL-RNA (Alu family) and Sn RNA genes, which doesn’t code for any proteins but some code for short proteins. 7 SL RNA is involved in regulating translation part of protein synthesis.
4.6 x 10^6bp x 0.30 x1.9/ 4.6x10^6 = 6.0 10^5bp
Alu family genes: copy numbers-1000 to 300 000.
tRNA genes: copy numbers-100 to 2000 or more.
r RNA genes: copy numbers-200 to 800 copies.
Sn/Sc RNA genes: copy numbers-100 to 500 or more.
Third component: ~ 4.6 x 10^6bp x 0.45 x 630 divided by 4.05 = 3.0 x 10^8 bp. It is non-repetitive DNA and has unique sequences; hardly they have any repetitive copies. All these DNA code for mRNAs for proteins and the said mRNAs (most of them) have substantial regions of non-coding sequences as Introns. Genes of these kinds do show some 3 to 4 or more copies.
4.6x10^6 x 0.45 x 630/ 4.05 = 3.0 x 10^8bp
Calculation of Chemical Complexity from Kinetic Complexity:
From the above experiment we know that the chemical complexity of the DNA used is 7x10^8 bp to conduct hybridization and determine Cot1/2. The KC (kinetic complexity) of the last component is 3 x 10^8 bp, which accounts 45 % of the total genome, so kinetic complexity multiplied by 45% gives the chemical complexity.
7x10^8 x .45 = 3.15 x 10^8
(Kinetic complexity) 3.15 x10^8 x (percentage of the genome) 0.45 = (chemical complexity) 6.6 x 10^8. This value is slightly lower than the actual CC (chemical complexity), because of experimental errors or deviations. Here the values given are mostly approximations.
Determining the Copy Numbers of each component or complexity:
Multiplying the CC with percentage of the genome it occupies and the product is divided by the kinetic complexity.
First: 7x10^8 x 0.25 / 340bp = 500 000 copies.
Second: 7x10^8 x 0.3 / 6.0 x 10^5bp= 350 copies.
Third: 7x10^8 x 0.45 / 3 x 10^8 bp = 1.05 copies.
Distribution of each of the components in some species:
|
Species |
Size |
First component |
Second component |
Third component |
|
E.coli |
4.6 x 10^6 bp |
Nil |
Nil |
100% |
|
C.elegans |
9.7 x 10^7 |
26 |
20 |
55 |
|
Drosophila |
1.65 x 10^8 |
14 |
14 |
72% |
|
M.musculus |
3.3 x 10^9 |
10 |
25% |
50 |
|
X.laevis |
3.1 x 10^9 |
5% |
41% |
54% |
|
N.tabaccum |
|
7% |
65% |
33% |
|
Arabidopsis thaliana |
1.19x10^8 |
41 |
24 |
35 |
C-value paradox, is not a paradox, it is a paradox in our heads, for our knowledge about the functional or non-functional part of the genome is so elusive (illusive!) and obscure in terms of the role played by each piece of DNA. By simplifying the genetic concepts looking at few organisms and their genome size and its functions, the puzzle is real and perplexing but not today. Organisms, with simple unicellular to multicellular, have an increase in their structural, as well as functional complexities; particularly regulatory functions and regulation of regulatory functions. Few years back it has been found (using data-base) that protein coding region of the DNA constitute only 1 to 1.2% of the human genome. The puzzle, what is rest of the DNA doing. Is it junk or useless DNA? It is now known except for Molecular fossil DNA and dysfunctional Transposons/ Retro transposons, the rest of the DNA code for a variety of RNA called non coding RNAs which have very important functions, and they can be called noncoding but functional RNAs, the number can be anywhere more than 18400?!. It beyond the imagination of Molecular Biologists, but it can be with time.
Leafing through the genomes of our major crop plants: strategies for capturing; unique information
Andrew H. Paterson; http://www.nature.com/
This graphic representation not same as the earlier graphics, shows four kinetic components, the first at the top is called Fold back (FB). It is the DNA that consists of palindromic sequences that self-anneal at rates independent of DNA. The graphics is the percent of ssDNA left as the function of cot Values. The other components are similar to those explained before; they are Highly Repetitive (HR), moderately repetitive (MR), and the last slow component (SL) (or unique sequences).
It is but natural to expect, with increase in organism’s structural and functional complexities the size of the genome has also also increased; however, this increase has taken 3.8 billion years of natural experimentation, standardization, selection, testing and stabilization. Molecular evolution at Genomic level is incredible and the process is still going on, where it will end; perhaps after another 4 to 5 billion years from now, that is time that our solar system also comes to an end?. The only part of the genome that doesn’t account for any function is highly repetitive DNA (not true) and to some extent some portion of the moderately repetitive DNA. Human genome consists of more than 40% of its genome as transposable and molecular fossil DNA. However now it is known there are several short sequences that produce small non coding RNAs such as sno RNA, Si/mi RNAs, sn RNA and scRNA, sRNA Rasi RNA, Tasi RNA, Xist RNA and many more of them not less than 74, yet they are functional RNAs. But looking at certain sequences and their location in chromosomal organization, it looks like they have some structural and functional roles. They are not there just because they happened to be there. The genomic contents of these first and second components account for substantial portion of the genome, which feature actually surprises every one. More surprising thing, that is contrary to the expectation, is with the total genomic size of humans being ~3 x 10^9 bp and by rough calculation based on the average size of mRNAs, the total number of protein coding genes expected to be present was about ~150, 000, but present calculation using genomic techniques, the number discerned is about 20000 to ~21000. For 20000 to 21000 genes the genome required, on the basis of 2000bp per gene or 5000bp per gene, is less than 4.4 x 10^6 or 1.5x10^8 bp long genomic DNA. But each of the eukaryotic genes has 5’UTR and 3’UTR and many-many introns whose size rages form little more than 100 to many thousand. Many of the introns spliced out are used as Sno RNAs. Yet there is lot of DNA to be unaccounted; what is it doing? This is another paradox. The answer in real terms is possible with more understanding of the genome, its structure and function. Look at genome complexity-called C-Value and compare with the number of Genes called G-value; this is called CDNA value paradox.
Using reassociation kinetics, it is also possible to get more information, such as what is the total number of functional genes found in a genomic DNA, what is the copy number of each gene expressed, What is total number of genes expressed as housekeeping genes in all tissues, which are they?, how many genes are expressed in tissue specific manner?. The said kind of information can be obtained by reassociation kinetics and also from High-density DNA microarray techniques. Reassociation techniques can be used to find out phylogeny and genetic relationship among the organisms, which is very important in understanding evolution and also the information can be used for improving plant and animal stock.
Determining the total number of structural genes:
The answer is simple. The haploid genome size, say Homo sapiens, 3.2x109 bp. Assume 45% percent of it is non-repetitive DNA, so its kinetic complexity is 3.375x108 bp. Assuming, simplistically, the average size of a Eukaryotic protein coding genes is 2000bp, total number of genes expected in the genome is =3.375x108 bp / 2000bp = 1.65x105 i.e. 165 000 genes which is really a large number. If the gene number is 165 000, how do you account for their structures and functions? Do human cells have so many functions to account for the number of genes deduced by calculations? But one has to consider how much of the DNA among the 165000 genes is non-coding, such as non-coding 5’ UTR and 3’ UTR terminal region and noncoding introns. Deducting this would give a fair amount of DNA sequence that actually accounts for protein coding region and other non-protein structural RNAs and ncRNAs. Human genome data suggest that the total number of expressed or structural genes are about 20000 to 21000. For that matter, calculations taking only protein coding regions i.e. only exons, the total percentage of DNA used for coding proteins is just 1% or 1.2% (3%); Matthew J. Hangauer, etal PLOS,equal contributor).
But the coding region consists of non-coding regions too. The number of proteins produced by the coded DNA exceeds the size of the DNA used; the rai-son d’etre is that a single pre-mRNA by alternative splicing can generate more number of proteins. One mRNA, DSCAM, can generate 38016 mRNAs by alternative splicing. This is considered as economical use of genomics. Besides protein coding part of the genome there are a large number of noncoding RNA genes (NcRNA), they produce functional small molecular weight RNAs. This accounts for another 1.2% of the genome or more. The intergenic transcripts including Linc RNA account for more than 10,000 ncRNAs (ENCODE). Here, by analyzing a large set of RNA-seq data, we found that >85% of the genome is transcribed, allowing us to generate a comprehensive catalog of an important class of intergenic transcripts: long intergenic noncoding RNAs (lincRNAs). We found that the genome encodes far more lincRNAs than previously known. A key question in the field is whether these intergenic transcripts are functional or transcriptional noise, equal; PLOS,
Matthew J. Hangauer, etal;
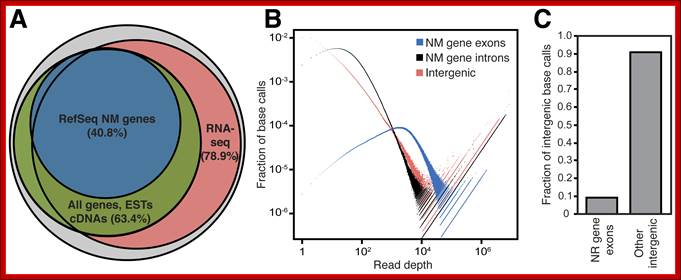
Pervasive Transcription of the Human Genome Produces Thousands of Previously Unidentified Long Intergenic Noncoding RNAs. PLoS Genet 9(6): e1003569. doi:10.1371/journal.pgen.1003569; Citation: Hangauer MJ, Vaughn IW, McManus MT (2013)
Sequence content of the human genome:
|
Class |
Copy numbers |
distribution |
% of human genome |
|
|
Single copy sequences |
~1 |
Euchromatin |
50% |
Protein-~1.2% NcRNA-~1% |
|
Low copy number sequences |
~2-20 |
Euchromatin, including Y chromo |
10% |
|
|
Moderately repeated NA |
~500 |
Scattered single copies |
25% |
|
|
High repeated DNA |
10K-500K |
Heterochromatin, but can be found in euchromatin |
15% |
|
This data from Introduction to human Genetics (Biology, B241)
Repetitive DNA
Highly repetitive DNA is often termed as Satellite DNA which can be isolated as separate band in buoyant density centrifugation. They are highly repetitive, where the repeat length varies for one few bp to hundred bps or more. If some sequences consisting of two to many 200 bp long and they are repeated tandemly, means one after another many times. The number of repeats can be few hundreds to few thousands. DNA-DNA hybridization kinetic studies show that a considerable amount of genomes in higher forms contain highly repetitive and moderately repetitive DNA.
Highly repetitive DNA:
The satellite DNA, when analyzed one finds the small and simple sequence repeats. Such repeats in strings can be identified by using a computer programme called suffix trees and /or suffix arrays. The highly repetitive sequences account for 10-15% of the mammalian genome, this includes tandem repeats.
The SAT- DNA can be apparent or cryptic, when it cannot be separated from the main band. Such sequences are generated by slippage processes and or illegitimate or unequal recombination and including translocation among the chromosomes. Such DNA can be identified by hybridization techniques.
The main band accounts for 92% and the satellite band accounts for 8%. This is not what we find in the repeated DNA analysis (whatever form of it is).
- It can account 10% to 40% in mammals and 50% in Drosophila virilis,
- They are organized in tandem repeats, 1000 or more repeats,
- They are found in large clusters and some in small clusters interspersed with non-repetitive DNA.
- They are inert and no function
- They are made up of short sequences that are repeated with identical copies or with little divergence.
- Size of the tandem repeats is polymorphic. Their size and sequence vary individually and number of repeats too varies.
- They are involved in synapsis and cross over in higher system.
- Genetic exchange among the SAT is high. They are formed and lost,
- These DNA are more or less AT rich GC poor,
- But GC rich heavy bands have been observed as sat DNA, they are found in cluster among the promoters of genes. There are at least 40,000 islands of GC clusters in human genome.
The AT rich regions are found in CEN region, peri-centromeric and Telomeric regions of the chromosomes. They can be differentially stained. Constitutive heterochromatin loci show satellite DNA. Such regions don’t contain genes, even if present hardly few and non-functional. The following are the kinds of SAT DNAs.
Alphoid DNA- 171bp repeats, found in all chromosomes.
Beta DNA- 68bp, found in CEN regions of chromosomes 1, 9, 13, 14, 15, 21, 22 and Y.
Satellite DNA1- 25-48bp, found in CEN and other constitutive HC regions.
Satellite DNA2- 5bp, found in most of the chromosomes.
Satellite DNA3- 5bp, found in most of the chromosomes.
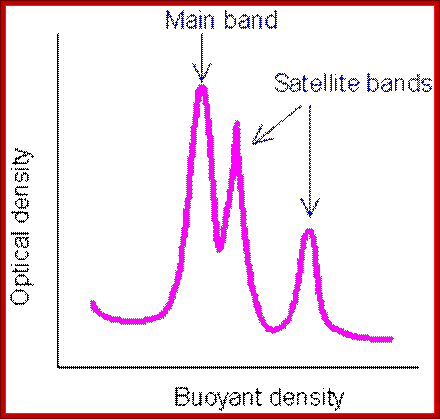
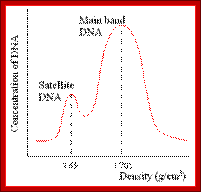
Illustration of satellite bands. By using buoyant density gradient centrifugation, DNA fragments with significantly different base compositions may be separated, and then monitored by the absorption spectra of ultraviolet light. The main band represents the bulk DNA, and the "satellite" bands originate from tandem repeats; http://www.web-books.com/
Minisatellite DNA:
They are made up of short sequences from 10-100bp and found in 1000 or more regions in Human genome.
Some SATs contain a core sequence “GGGCAGGAXG” repeated many times. These act as hotspots for chromosomal exchange that can generate more minisat with wide distribution and numbers. The most mutable ‘sat’ identified is CEB1 (D2S90).
Consists of GC rich segments and the repeats vary from 10-100bp long. The variants are intermingled thus it can be used for DNA turnover process. Crossing over between such repeat sequences can lead to increased and decreases lengths.
GGCAGGG
X
GGGCAGGG
= GGGCGGCAGGG + AGGTT
Variation among the minisats can be used for genetic fingerprinting (discovered by A.R Wyman and R. White). Alec Jeffrey used this for fingerprinting; they are also used as genetic markers for linkage analysis and population studies.
They are also implicated in gene expression, -level of expression, alternate splicing and /or imprinting.
Some minisats act as fragile sites and hyper mutable with plus or minus 0.5 to 20%. Minisatellite variant repeat mapping (MVR) by PCR is used to determine the turnover of these segments. Flanking regions are involved in recombination.
Telomeric satellites: They are exclusively found in chromosomal ends with six base repeats TTAGGG TTAGGG TTAGGG, species specific found in telomeric and sub-telomeric region. The repeats very often expand and contract and repeats can be 500 to 10000 or more.
They are termed as Variable Number of Tandem repeats (VNTR); such sequences are used for DNA finger printing. The numbers vary and scattered. Minisatellite show high variability
People have developed primers for them; by amplifying them one get a band pattern which is specific to a particular species and specific individual.
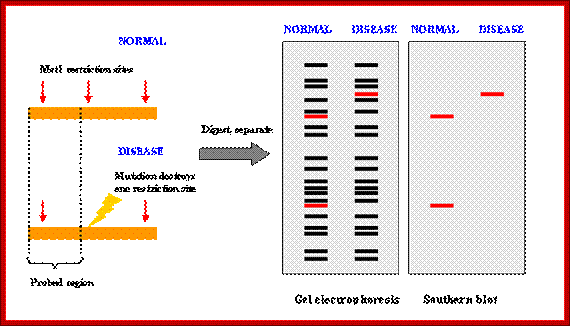
How RFLP works. SNPs or INDELs can create or abolish restriction endonuclease (RE) recognition sites, thus affecting quantities and length of DNA fragments resulting from RE digestion. http://www.ncbi.nlm.nih.gov/

Genotyping;http://www.ncbi.nlm.nih.gov/
Developing RFLP probes
- Total DNA is digested with a methylation-sensitive enzyme (for example, PstI), thereby enriching the library for single- or low-copy expressed sequences (PstI clones are based on the suggestion that expressed genes are not methylated).
- The digested DNA is size-fractionated on a preparative agarose gel, and fragments ranging from 500 to 2000 bp are excised, eluted and cloned into a plasmid vector (for example, pUC18).
- Digests of the plasmids are screened to check for inserts.
- Southern blots of the inserts can be probed with total sheared DNA to select clones that hybridize to single- and low-copy sequences.
- The probes are screened for RFLPs using genomic DNA of different genotypes digested with restriction endonucleases. Typically, in species with moderate to high polymorphism rates, two to four restriction endonucleases are used such as EcoRI
, EcoRV, and HindIII. In species with low polymorphism rates, additional restriction endonucleases can be tested to increase the chance of finding polymorphism (www.ncbi,nlm,nih.govt).
PCR-RFLP
Isolation of sufficient DNA for RFLP analysis is time consuming and labor intensive. However, PCR can be used to amplify very small amounts of DNA, usually in 2-3 hours, to the levels required for RFLP analysis. Therefore, more samples can be analyzed in a shorter time. An alternative name for the technique is Cleaved Amplified Polymorphic Sequence (CAPS) assay. http://www.ncbi.nlm.nih.gov/
Micro satellite DNA:
Micro satellite DNA- is still shorter than minisatellite DNA. Mammalian genomes contain more than 10% of this DNA,
These are simple sequence repeat elements (SSR) or short tandem repeats (STR). Human genome consists of at least 30,000 micro satellite loci. They consist of 1-4 bp, repeated tandemly 10 to 1000 times or more. Example CA, TCG, GGC, TTA, TTCC, TCTA, TAGG etc. Human genome contains at least 10,000 published STR sequences-extensively used in genetic profile studies. The CEN region contains highly repetitive sequences and they are classified into SAT I, II, III, IV and alpha types. Most of them are located in centromeric and para-centromeric and constitutive heterochromatic regions.
Micro satellite DNA became popular in 1990s; now it is used in forensic studies, where they use tetrameric or pentameric sequences for 4 to 5 repeats or more. They give error free data, but longer repeats do produce error.
Capillary electrophoresis, Acrylamide gel electrophoresis and Agarose gel electrophoresis are used to detect labeled bands (radioactive or fluorescent or even non-radioactive components -silver chloride).

Inter simple sequence repeat PCR. (ISSR-PCR):
These are intergenic micro satellite regions i.e. found between micro satellites. These sequences are used for amplifying micro satellite segments and very useful in Genetic fingerprinting. They are versatile markers, for population studies. They can be used for comparison between species and among the species for variation identity.

Principles and protocol; http://realtimepcr.dk/
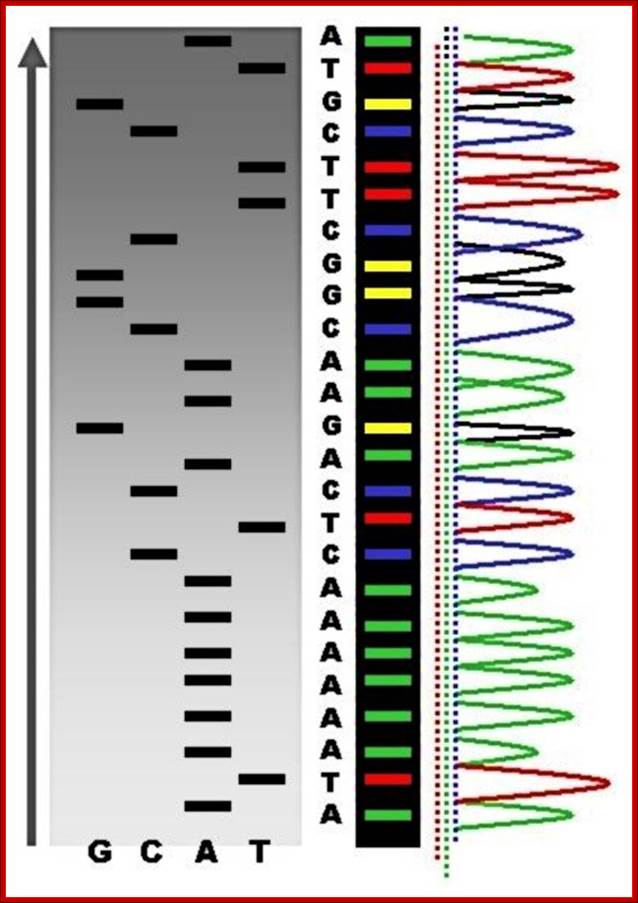
PCR machines used for DNA sequences determination and gel showing DNA sequences with color codes; Recent DNA sequence technologies are very sophisticated, and quick to analyze and cheaper cost for the user; ttp://en.wikipedia.org/
Genetic profile;
Every person's DNA is so unique that it can be used for identification.
When an individual's DNA is profiled only short sections of Non-coding DNA are used. Our genome contains simple repetitive sequences that are scattered throughout our 46 chromosomes. Some of these repetitive sequences are only 2–6 base pairs in length and are called microsatellites or short tandem repeats (STRs). Each person has microsatellites that vary in length and can be used to distinguish individuals. This principle was used to develop DNA profiling for forensic investigations and tracing ancestry. Some countries, including the United Kingdom and New Zealand, have established national DNA data bases that operate by targeting approximately 10–13 STRs. This ensures that it would be highly unlikely to get two identical results from two people.
Profiling can be carried out quickly and efficiently by making multiple copies of DNA segments. This is done by a technique known as PCR.
Paternity testing is a form of DNA profiling. The DNA of the man who may be the father of a child is compared to the child's DNA and to that of the mother.
An example of the DNA bands from a paternity test is shown below. The standard band provides a control or comparison with which to compare the samples.
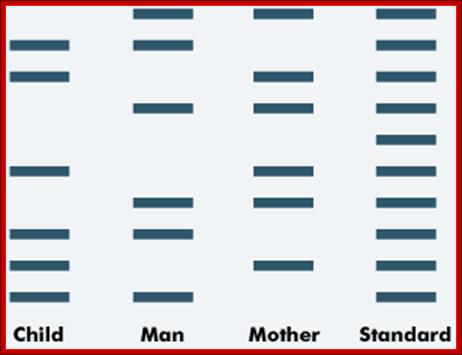
Paternity testing; http://tle.westone.wa.gov.au/
The methods of DNA sequencing
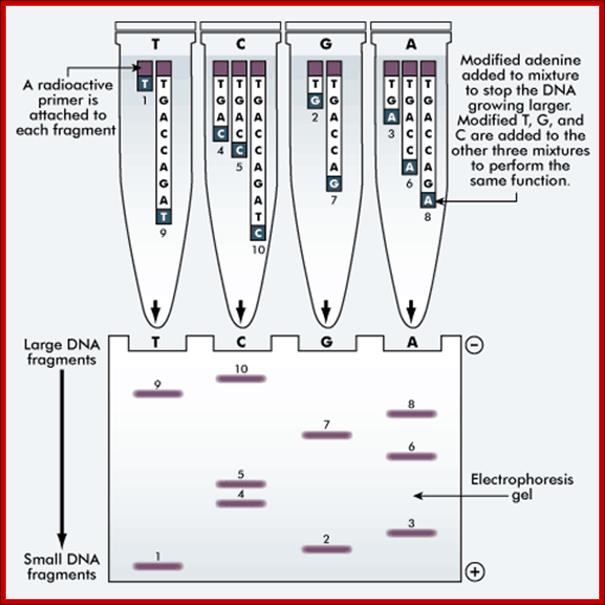
The 'reading' of a DNA molecule is achieved by either a manual or an automated method of sequencing. The Sanger’s method (outlined below) is a manual method. Both methods use electrophoresis to sort the DNA into a format that can be analyzed.
The Sanger method of DNA sequencing;
This method uses the same DNA sample. A separate sequencing reaction is carried out for each nucleotide, ie A, T, G and C.
A radioactive primer is attached to each fragment. The modified adenine is added to the mixture to stop the DNA from growing larger. The modified T, G and C are added to the other three mixtures to perform the same function.

The DNA sequence of an individual can be determined and a printout of the base sequence provided using the automated method. The result can be compared with other DNA sequences. http://tle.westone.wa.gov.au/DNA analysis techniques of the past 50 years have been used to establish species similarity on a base-pair level. Given two species, the degree of similarity between their DNA is closely mirrored by similarity in morphology. For example, the DNA of humans and chimps is 98% identical. Modern sequencing techniques are so advanced that family's can submit their DNA to determine which countries their great-grandparents migrated from. http://www.listoid.com/
Genomic DNA contains simple sequence repeats (SSR) or simple tandem repeats -STR. The SSR repeat may contain (CA) n and (AT) n, generated by slipped DNA replication; SSR can be 3% of the human genome.
The most common micro sat sequence is CA(n), where n is variable.
CA sequences are very frequent in human genome. Present at every few thousand bp region. There are many alleles because of variations.
This feature is very important in determining paternity and useful in population studies and can be used as molecular markers.
Such sequences are found in nuclear DNA and also in organelle DNA.
They are co dominant, neutral, they are used in kinship studies and also in population studies. They show high rate of mutation for during replication they go through slippage and during recombination events. Using flanking regions as primers one can use PCR methods to identify the repeat patterns as bands. Even random primers are used for genetic finger printing.
Moderately Repetitive DNA:
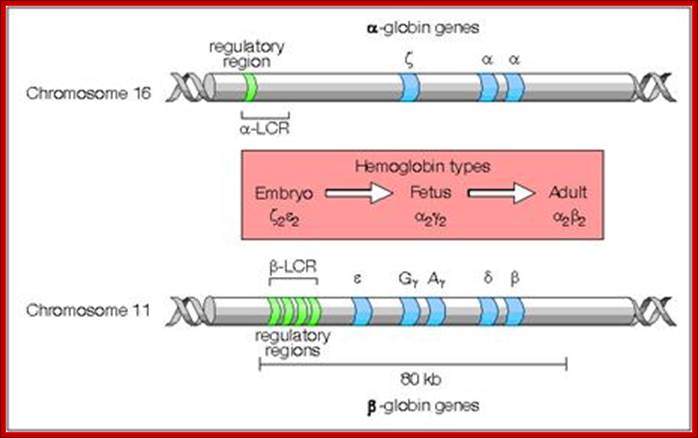
Most of the rRNA genes, tRNA genes, histone genes, globin (Hg) genes, organized into tandem repeats, Sn RNA genes, snoRNA genes; the above form gene families.
Numbers range from 50 -200 or more; They are derived from duplication, unequal recombination and variation; some are just duplicated to the requirements. rRNA, snRNA, scRNA, tRNA, snoRNA genes are moderately repetitive DNA; they constitute 20.4% of Hu.genome (Hu=Human).
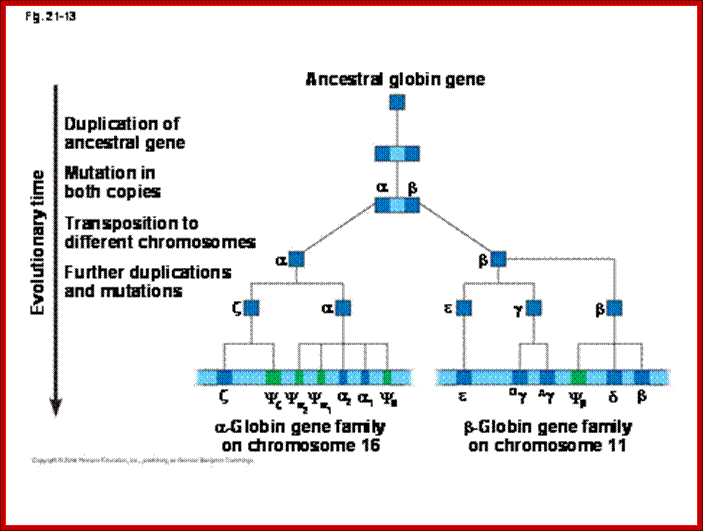
Genomes and Evolution of genes with novel functions; http://www.mrschamberlain.com/
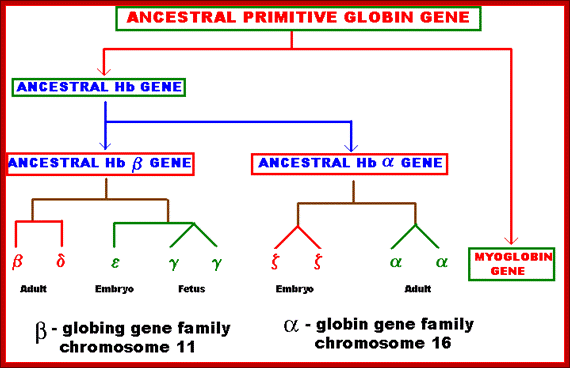
Several types of genes occur in similar but not identical forms. These forms produced by the process of gene duplication. The copies diverged in function. These gene families include globin genes, Ig genes, chorion protein genes in insect egg shell and Drosophila heat shock genes. For example primitive globin gene is initially converted into two globin genes by gene duplication process namely hemoglobin gene and myoglobin gene. These two gene sequences found to be similar but not identical. Because of this, these genes have similar function. Similarly hemoglobin gene undergoes further duplication to form types like Alpha, Beta, Gamma, Delta, Epsilon and Zeta. Eventhough hemoglobin with different combination of polypeptides has oxygen transport function; they differ in their affinity to oxygen. This is the reason for the existence of different polypeptide chain types. http://biosiva.50webs.org/
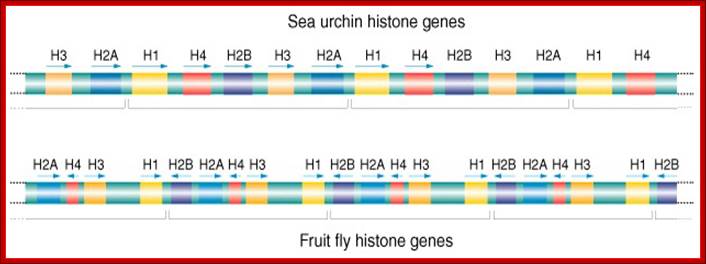
Histone genes- Tandem Repeats; Multiple Genes; http://www.cas.miamioh.edu/
Rate of duplication take one million years to duplicate 1% of the genes, 0.1 of them to diversify take million years, silencing of their copies may take another 4 million years. The rRNA, tRNA genes account for substantial amount of the genome. Some of the other DNA sequences, 100 to 6000bp are repeated several hundred times or more. They form distinct class of elements called mobile elements.
Mobile Elements;
Mobile genetic elements (MGE) are a type of DNA that can move around within the genome. They include:Transposons (also called transposable elements);
· Plasmids
· Bacteriophage elements, like Mu, which integrates randomly into the genome
The total of all mobile genetic elements in a genome may be referred to as the mobilome. Barbara McClintockwas awarded the 1983 Nobel Prize in Physiology or Medicine "for her discovery of mobile genetic elements".en.wikipedia.org.
Mobile genetic elements play a critical role in the spread of virulence factors, such as exotoxins and exoenzymes, amongst bacteria. Strategies to combat certain bacterial infections by targeting these specific virulence factors and mobile genetic elements have been proposed
Such gene transfers an happen due to Horizontal gene transfer-transfer between organisms and lateral gene transfer-transmission from the parental generation to off springs via sexual or asexual reproduction. HGT is an important factor in genetic changes among organisms and evolution.
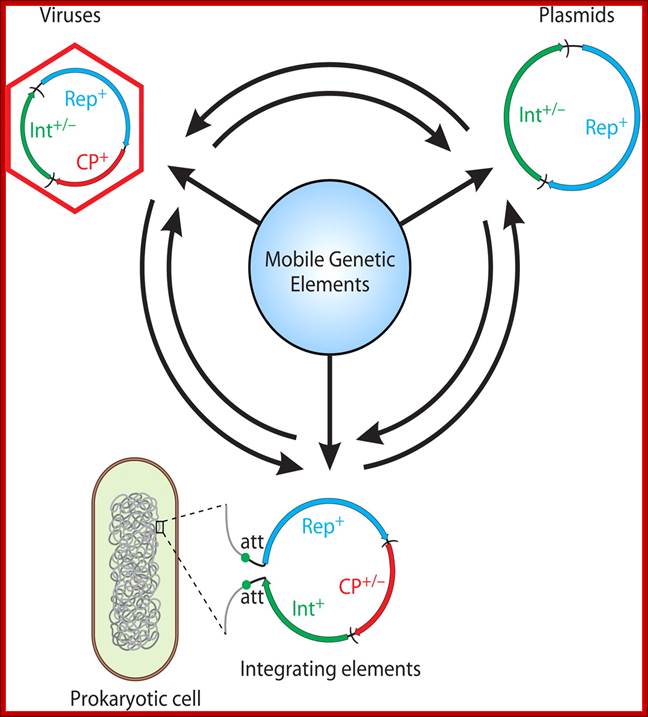
Dynamics within the prokaryotic Virosphere; http://mmbr.asm.org/
Genomic relationship between different groups of mobie elements . Genoic modularity is characteristic of different types of mobile genetic elements and the mechanism of transfer and transmission.
They are characterized by their ability to transpose from one site to the other. They were first discovered by Barbra McClintock, in early 1940-1948 in corn plants expressed phenotypically in the form of colored patches on corn grains. The author called them as Activator elements (Ac) and Dissociation elements (Ds) elements. This discovery is many decades earlier to the finding of transposable elements. She observed deletions, insertions, and translocations caused by these Transposons. She was not a molecular biologist, but her work was recognized all over the world and one should appreciate how and why genetics has the basics of molecular biology. Such changes resulted in observable color changes in the endosperms of corn grains, so a single cob to contain several colored fruits, here one ovary develops into one fruit and all of them organized on the same axis. Nearly 50% of the maize genome consists of such transposable elements. Her work was recognized and she was awarded with a Nobel prize in 1983; late but worth it in diamonds.
Graphic analysis of the human genome:

https://lookfordiagnosis.com; http://mhuel.wordpress.com/
http://commons.wikimedia.org/and http://www.polypompholyx.com/
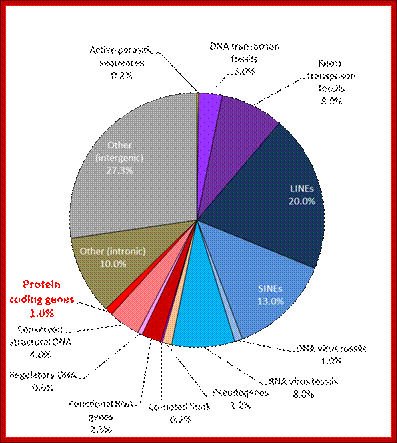
In 2001, a rough draft of most of the human genome was published by the Human Genome Consortium. A break-down of the data supports the suggestion that much of the genome of humans (and by implication, even more of the genome of salamanders) is “junk”:
Only one or two percent of the human genome codes for the proteins that construct your body. This DNA is transcribed to messenger RNA, which is then translated to protein, a process whose details have been used to torture undergraduate students for many decades.
Another two or three percent of your DNA codes for functional RNA molecules, which are vital components of the machinery that translates messenger RNA into protein. A few more percent are structural, forming the ends of your chromosomes, or providing the anchor points needed to pull chromosomes around when your cells divide. One percent or so is of obvious regulatory function, helping to bind proteins that turn your genes on and off as required.
So, all in all, about 10% of your genome (the reddish segments of the pie chart above) is of well-known, long-established, and unambiguous function. The other 90% seems to be superfluous “junk”. Despite all the furore generated by the ENCODE project, nothing written above is news. Only about 1% of your genome directly encodes proteins, but it’s been well-known for a long time that at least 10% of your genome is “functional” by a slightly broader but uncontroversial definition. W3.Polypompholyx.com; http://mhuel.wordpress.com/
The human genome is organized into chromosomes (very large linear DNA molecules contained within the cell nucleus) and the small mitochondrial DNA (a comparatively tiny circular DNA molecule). Basic information about these molecules and their gene contents are provided in the following table. (Data source: Ensembl genome browser release 68, July 2012).

https://www.boundless.com
The above figure summarizes the physical organization and gene content of the human reference genome, with links to the original analysis, as published in the Ensembl database at the European Bioinformatics Institute (EBI) and Wellcome Trust Sanger Institute. Chromosome lengths were estimated by multiplying the number of base pairs by 0.34 nanometers, the distance between base pairs in the DNA double helix. The number of proteins is based on the number of initial precursor mRNA transcripts, and does not include products of alternative pre-mRNA splicing, or modifications to protein structure that occur after translation.
The number of variations is a summary of unique DNA sequence changes that have been identified within the sequences analyzed by Ensembl as of July, 2012; that number is expected to increase as further personal genomes are sequenced and examined. In addition to the gene content shown in this table, a large number of non-expressed functional sequences have been identified throughout the human genome (see below). Links open windows to the reference chromosome sequence in the EBI (E), NCBI (N), or UCSC (U) genome browsers. The table also describes prevalence of genes encoding structural RNAs in the genome.
MiRNA, or MicroRNA, functions as a post-transcriptional regulator of gene expression. Ribosomal RNA, or rRNA, makes up the RNA portion of the ribosome and is critical in the synthesis of proteins. Small nuclear RNA, or snRNA, is found in the nucleus of the cell. Its primary function is in the processing of pre-mRNA molecules and also in the regulation of transcription factors. SnoRNA, or Small nucleolar RNA, primarily functions in guiding chemical modifications to other RNA molecules.
Coding vs. noncoding DNA.
The content of the human genome is commonly divided into coding and noncoding DNA sequences. Coding DNA is defined as those sequences that can be transcribed intomRNA and translated into proteins during the human life cycle; these sequences occupy only a small fraction of the genome (<2%). Noncoding DNA is made up of all of those sequences (ca. 98% of the genome) that are not used to encode proteins.
Some noncoding DNA contains genes for RNA molecules with important biological functions (noncoding RNA, for example ribosomal RNA and transfer RNA). The exploration of the function and evolutionary origin of noncoding DNA is an important goal of contemporary genome research, including the ENCODE (Encyclopedia of DNA Elements) project, which aims to survey the entire human genome, using a variety of experimental tools whose results are indicative of molecular activity.
Because non-coding DNA greatly outnumbers coding DNA, the concept of the sequenced genome has become a more focused analytical concept than the classical concept of the DNA-coding gene.[5][6]
Coding sequences (protein-coding genes)
![]()
Human genes categorized by function of the transcribed proteins, given both as number of encoding genes and percentage of all genes. https://www.boundless.com
Protein-coding sequences represent the most widely studied and best understood component of the human genome. These sequences ultimately lead to the production of all human proteins, although several biological processes (e.g. DNA rearrangements and alternative pre-mRNA splicing) can lead to the production of many more unique proteins than the number of protein-coding genes.
The complete modular protein-coding capacity of the genome is contained within the exome, and consists of DNA sequences encoded by exons that can be translated into proteins. Because of its biological importance, and the fact that it constitutes less than 2% of the genome, sequencing of the exome was the first major milepost of the Human Genome Project.
The number of protein-coding genes within the human genome remains a subject of active investigation. A 2012 analysis of the human genome based on in vitro gene expression in multiple cell lines identified 20,687 protein-coding genes.[3] Although the estimate of the number of protein genes has varied widely, ranging up to 2,000,000 in the late 1960s,[8] several researchers pointed out in the early 1970s that the estimates of mutational load from deleterious mutations placed an upper limit of approximately 40,000 for the total number of functional loci (this includes protein-coding and functional non-coding genes).[9]
The number of human protein-coding genes is not significantly larger than that of many less complex organisms, such as the roundworm and the fruit fly. This difference may result from the extensive use of alternative pre-mRNA splicing in humans, which provides the ability to build a very large number of modular proteins through the selective incorporation of exons
Protein-coding genes are distributed unevenly across the chromosomes, with an especially high gene density within chromosomes 19, 11, and 1 (Table 1). Each chromosome contains various gene-rich and gene-poor regions, which may be correlated with chromosome bands and GC-content[citation needed]. The significance of these nonrandom patterns of gene density is not well understood.[10]
The size of protein-coding genes within the human genome shows enormous variability (Table 2). For example, the gene for histone H1a (HIST1HIA) is relatively small and simple, lacking introns and encoding mRNA sequences of 781 nt and a 215 amino acid protein (648 nt open reading frame). Dystrophin (DMD) is the largest protein-coding gene in the human reference genome, spanning a total of 2.2 MB, while Titin (TTN) has the longest coding sequence (80,780 bp), the largest number of exons (364), and the longest single exon (17,106 bp). Over the whole genome, the median size of an exon is 122 bp (mean = 145 bp), the median number of exons is 7 (mean = 8.8), and the median coding sequence encodes 367 amino acids (mean = 447 amino acids;
Human proteins categorized by function, given both as number of encoding genes and percentage of all genes (WIKI).
Genomic and transcriptional characterization of cancer cell lines
A
panel of the most frequently mutated cancer genes was sequenced to base-pair
resolution across all coding exons for each gene by capillary sequencing in our
panel of human cancer cell lines, and which formed the basis for the cell lines
chosen for this drug screen. The presence of commonly rearranged cancer genes
(e.g. BCR-ABL, MLL-AFF1 and EWS-FLI1) was determined across the drug screen
cell line panel by the design of breakpoint-specific sequence primers that
enabled the detection of the rearrangement following capillary sequencing.
Analysis of microsatellite instability (MSI) was carried out according to the
guidelines set down by "The International Workshop on Microsatellite
Instability and RER Phenotypes in Cancer Detection and Familial
Predisposition" workshop. Samples were screened using the markers BAT25,
BAT26, D5S346, D2S123 and D17S250 and were characterised as MSI if two or more
markers showed instability. Total integral copy number values across the
footprints of the cancer genes were determined from Affymetrix SNP6.0
microarray data using the 'PICNIC' algorithm to predict copy number segments in
each of the cell lines. For a gene to be classified as amplified, the entire
coding sequence must be contained in one contiguous segment defined by PICNIC,
and have a total copy number of eight or more. Deletions must occur within a
single contiguous segment with copy number zero. For gene expression analysis,
RNA was extracted from each cell line using a standard Trizol protocol and
hybridized to the HT-HGU122A Affymetrix whole genome array. Normalised gene
expression intensities were generated using the Robust Multi-Array Average
(RMA) algorithm. A complete description of the characterization of our cancer
cell lines collection is available from the Cancer Genome Project webpages (http://www.sanger.ac.uk/genetics/CGP/CellLines;![]() ).
http://www.cancerrxgene.org/
).
http://www.cancerrxgene.org/
http://www.mrschamberlain.com/

http://www.theologyonline.com/
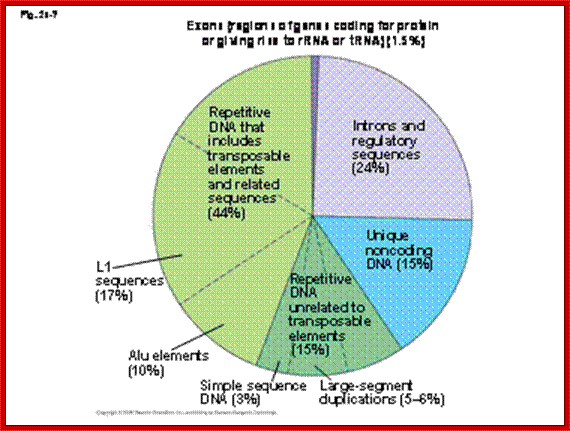
Frequency of different types of DNA elements
"Junk" DNA and why we know it exists in the human genome or Why ENCODE is wrong - December 9th, 2013, 09:30 PM
This
topic has come up from various posters in various threads so I figured it
deserved it's own thread. This will be unfortunately technical, but it can't
really he helped.
The concept of "junk DNA" has had a convoluted history. The basic
idea is that many organisms (including humans) can have relatively large
amounts of DNA that are not necessary for the ordinary functioning of the
organism, but don't seem to be causing any harm. There has been some confusion
in the popular press about the term - that it originally applied to all DNA
that does not code for proteins. That is untrue.
The concept grew out of something known as, The C-Value Enigma. C-Value
referring to the amount of DNA a particular organism contains. And what most of
us would expect is that more complicated organisms should have more DNA (and
presumably more genes) than simple organisms. This is one of the few ideas
"evolutionists" and creationists would probably share. And it was a
great idea (like so many) until it ran into data.
What scientists found instead is incredibly wide variation in genome size. Closely
related organisms can have vastly different genome sizes. Size is measured in
base pairs meaning one rung of the famous twisty ladder of DNA. Each is a
single pair of DNA letters, ATGC.
Human: 3.2 Billion Base Pairs;Mouse: 2.7 Billion Base Pairs; Puffer Fish: 0.36
Billion Base Pairs; Marbled Lungfish: 133 Billion Base Pairs; Ostrich: 2.1
Billion Base Pairs;Black Chinned Hummingbird: 0.88 Billion Base Pairs; Canopy
Plant: 150 Billion Base Pairs; Corn/Maize: 2.8 Billion Base Pairs
Arabidopsis (model plant): 0.135 Billion Base Pairs.
It would make sense that a marbled lungfish doesn't really NEED a genome 40
times larger than the human genome. Nor does a canopy plant NEED a genome over
1000X larger than that of Arabidopsis. And it's incredible to think a pufferfish can get away with a genome one
tenth the size of the average vertebrate. And there are single celled organisms
that have genomes larger than any of the ones listed above. Why would they
"need" such large genomes?
The answer would seem to be that some species have "extra" DNA, more
than is necessary. But where does this apparently "extra" DNA come
from?
Scientists then started to learn about the *composition* of genomes. And
instead of being filled with orderly genes, and other such sequences, scientists
found large regions of repetitive DNA (meaning the same bases were used over
and over) and not only was it repetitive it looked like pieces of viruses and
transposons.
Transposons are pieces of
DNA that can copy themselves and multiply in a genome. These are often called
"selfish DNA" since they can propagate through a genome and normally
provide no adaptive advantage to the organism that contains them.
In the human genome these sequences are a relatively large proportion of the
genome.
In the figure below: Exons being coding regions of genes, introns being
important to regulating genes. Unique non-coding DNA probably serving a
currently unknown purpose. But transposon related repetitive DNA makes up a
whopping 44% of the total human genome.w3. theology line.
1. You might say, how do we know that the "extra"
DNA isn't essential? For one thing, we have created mice in the laboratory that were missing large sections of this DNA, with
no ill effects.
2. Secondly there is variation within human
populations as to how much of this repetitive DNA they have. It's actually the
basis of genetic testing for identification purposes, paternity, crimes etc.
3. Thirdly from an evolutionary perspective,
DNA that is important to an organism should be very similar or *conserved*
between closely related organisms. When some parts of a genome are clearly very
close, whereas other parts of the genome do not bear close resemblance at all,
and usually these are non-coding repetitive regions that appear to be derived
from transposons. Encode analysis. In maize transposons make up 85% of the
genome.
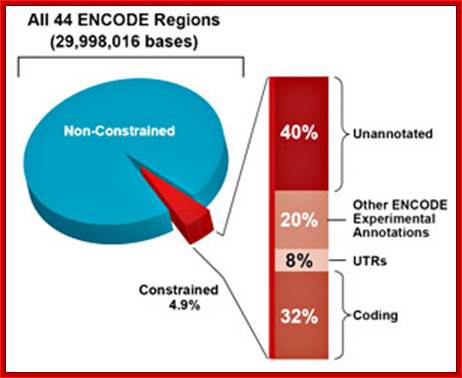
One of the key lessons that ENCODE has taught us is that not all functional genomic elements are evolutionarily constrained and that many evolutionarily constrained elements perform functions that are currently unknown; ENCyclopedia Of DNA Eelement (ENCODE) published in Genome Research; http://www.sanger.ac.uk/

MicroRNA target Transposons in plant reproductive cell,protecting against Genome damge; Grain phenotypic effect due to Transposons. http://www.bioquicknews.com/
Many of the moderately repetitive DNAs are insertional sequences, which are Transposons, which are in fact Mobile elements for they jump from one site to the other in the genome. Such elements or sequences are classified as insertional elements or Transposons. They are found in both bacteria and eukaryotes.
|
IS |
Length |
End repeats |
Target site/size |
Copy no. |
|
IS1 |
768 |
20/23 |
9 |
5-8 |
|
IS2 |
1327 |
32/41 |
5 |
5 |
|
IS3 |
1258 |
39/19 |
3 |
5 |
|
IS4 |
1426 |
16/18 |
11-14 |
1or2 |
|
IS5 |
1195 |
15/16 |
4 |
Abundant |
|
IS10R |
1329 |
17/22 |
9 |
|
Bacterial Insertional Elements (IS):
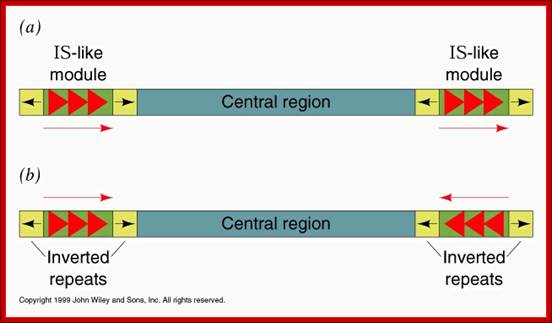
They have simple structural features, consisting of 700-1500bp long DNA flanked by 20-40bp long inverted repeat sequences. The central segment codes for an enzyme called Transposase that catalyses transposition. Transposase cuts the entire segments and transposes to another site. There is no sequence specificity for insertion. This jumping into the middle of functional gene causes genetic defect. There many types of IS elements in bacteria.
|
|
|
Bacterial Transposons: They are similar to insertional elements. The central DNA segment 5000bp or more, codes for more than one product and flanked by short inverted repeat sequences. The central DNA may contain Transposase, resolvase and possibly an antibiotic resistance gene.
|
||||||||||||||||||||||||||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||||||||||||||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||||||||||||||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||||||||||||||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||||||||||||||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||||||||||||||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||||||||||||||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||||||||||||||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||||||||||||||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||||||||||||||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||||||||||||||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||||||||||||||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||||||||||||||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||||||||||||||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||||||||||||||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||||||||||||||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||||||||||||||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||||||||||||||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||||||||||||||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||||||||||||||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||||||||||||||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||||||||||||||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||||||||||||||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||||||||||||||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||||||||||||||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||||||||||||||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||||||||||||||||||||||||||
Composite Transposons:
These Transposons contain DNA segments flanked by IS elements. In this, transposition includes central as well as IS elements.
|
Transposon |
Central (bp) |
IS (bp) |
Marker |
|
Tn5 |
5700 |
Is50 |
kanamycin |
|
Tn9 |
2638 |
IS1 |
chloramphenicol |
|
Tn10 |
9300 |
IS10 |
tetracycline |
|
|
|
|
|
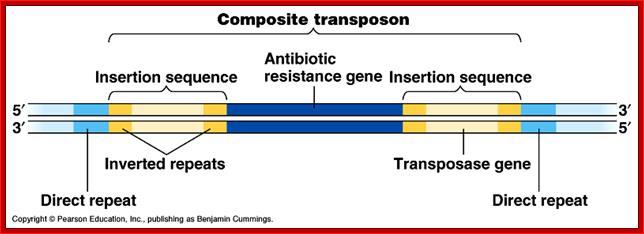
http://www.zo.utexas.edu/;http://www.freya.u-net.com/

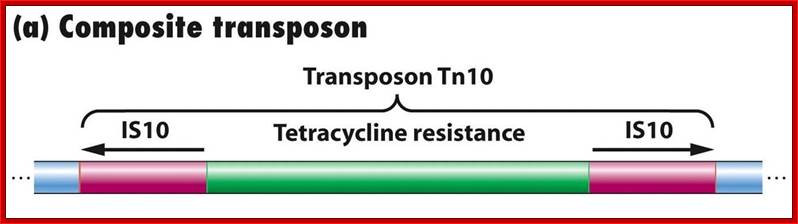
General structure of insertion sequences and transposons. Flanking direct repeats (FDRs) are shown as green triangles, inverted repeats (IRs) are red or purple triangles, insertion sequences (ISs) are yellow boxes with red triangles at the end, and other genes are boxes of different colors. The boxes and triangles include both strands of duplex DNA. DNA outside the FDRs is shown as one thick blue line for each strand. Tn5 has an IS50 element on each side, in an inverted orientation. Transcripts are shown as curly lines with an arrowhead pointing in the direction of transcription. The neoR gene for Tn5 is composed partly of the leftward IS (ISL) and partly of other sequences (included in the blue box). The transposase for Tn5 is encoded in the rightward IS (ISR).
Interestingly phages such as ‘mu’ gets inserted randomly and
transposes by replicative mechanism, thus it acts like a transposon.
Transposition mechanism:
Simple transposition:
This is also called direct or conservative mechanism, where the Transposase produced cuts the flanking region leaving double stranded break; in the process the original molecule is damaged. The Transposase cuts at another site creating staggered cut and the segment is ligated and then repaired, resulting in the duplication of ends.
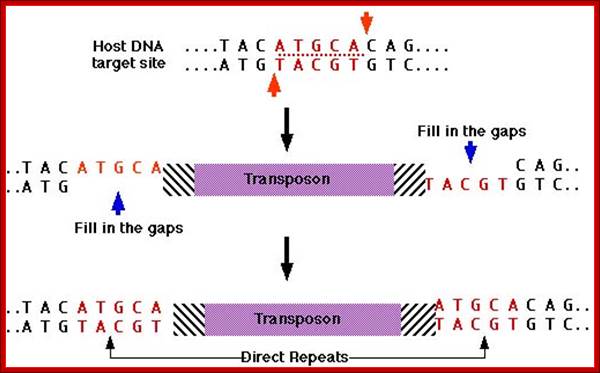
http://users.rcn.com/
Replicative Transposition:
In this process the transposon segment is cut on either side of the transposon and also produces another cut at another site. Here a cleavage result is staggered cuts. Then the cut segment is integrated in the next site.
Class I Mobile Elements-
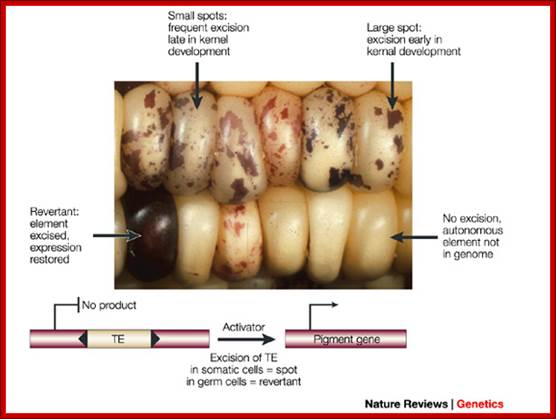
Corn color concepts: http://biologicalexceptions.blogspot.com/
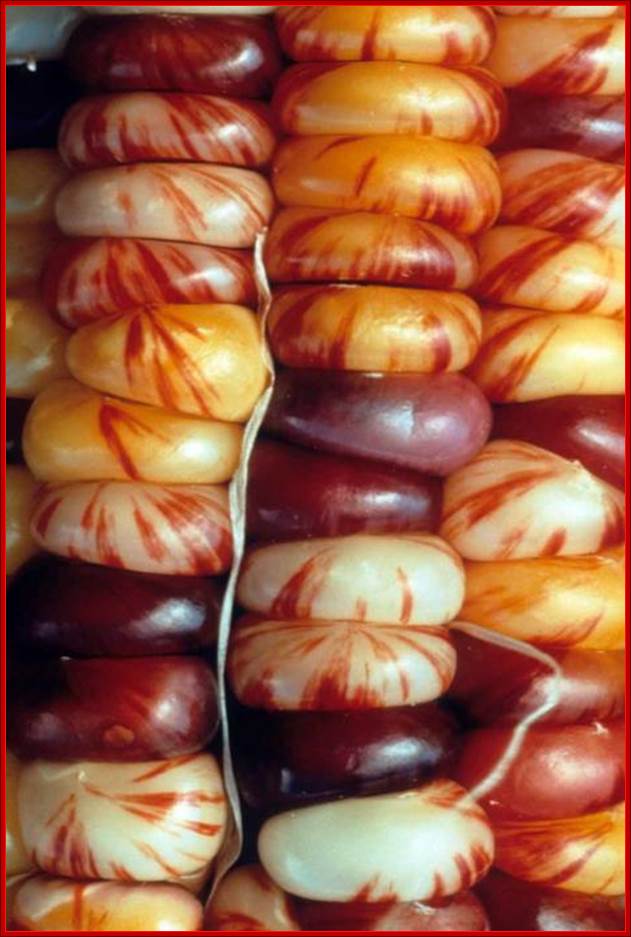
The corn cobs with grains color with pattern due to Transposon insertions. http://magpiesmiscellany.wordpress.com/
Retrotransposons:
They are genetic elements can amplify themselves and they are ubiquitous in higher genomes.
They are derived from retro viruses or eukaryotic RNAs, by reverse transcription and insertion. They are the subclass of Transposons.
They are abundant in plants accounting to a large portion of the genome, ex Maize 49-78% is Retrotransposons, wheat contain 90% repeat sequence and 68% Retrotransposons. Mammalian genome contains 48% of transposons. Around 42% of the human genome consists of Retrotransposons. And DNA transposons account for 2-3%. This can be said as that for every gene there are 3 types of Retrotransposons.


Brilliant colors and Barabara McClintock; Ty1 and L11http://magpiesmiscellany.wordpress.com/
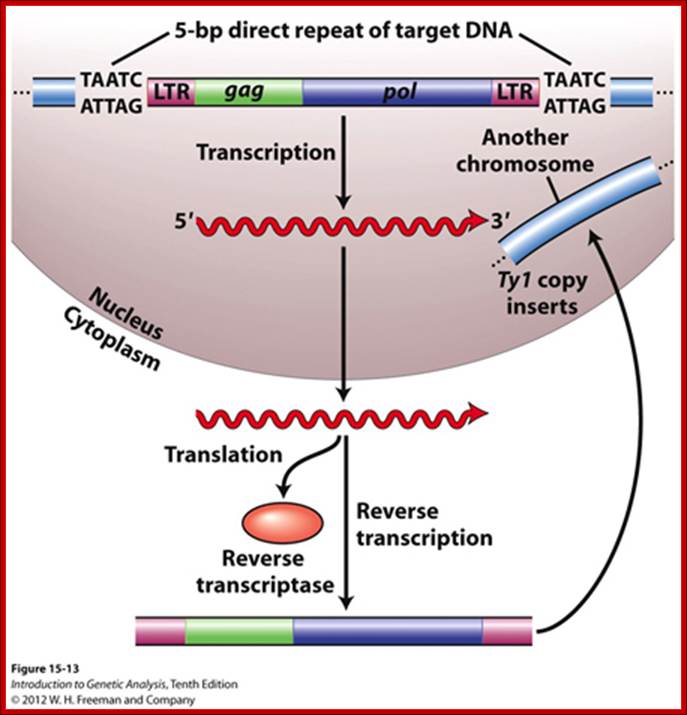
The Copia, Ty1 and L1 are derived from retroviral insertions:
![]()
Biol202 Genetics http://www.discoveryandinnovation.com/BIOL202
Retrotransposons use replicative mode of transposition via RNA. Thus they increase copy numbers rapidly and also increase the genome size. Retrospons induce mutation and such mutations are found to be stable. Transposition and survival requires contribution from the Retrospons or Retrotransposons and host factors.
They have evolved and coexist for million years of plant history. However majority of them have lost functions because of mutations and no more able to transpose and existing as dead fossils, thus contribute to high genome size. Increase in genomic content has some advantages, for they act as buffers against point mutations
Types of Retrospons:
LTR Containing Retrotransposons:
They contain direct long terminal repeats containing U3-R and U5 segments made up of several hundred base pairs long. The size of the Retrospons is ~5kbp to 6kbp or more. They use polymerase, Integrase to copy and insert.
U3-R-U5------------------------U3-R-U5
They account for 8% of the human genome. They are found in million copies in plants with large genomes.
The Ty element lacks the env ("envelope") gene, which encodes the retroviral protein capsid. Without a capsid, the retrotransposon remains just a naked bit of DNA jumping around the genome. Ty elements are sometimes considered primitive retroviruses; http://www.bio.miami.edu/
The diverse array of transposable elements (TEs) can be classified using several criteria, such as the requirement for a reverse-transcription step to transpose. TEs that require a reverse-transcription step are called retro transposons, or type I elements. They can be divided into two types, on the basis of the presence or absence of direct repeats at the ends of the element called long terminal repeats (LTRs). LTR retrotransposon proteins pol and gag are closely related to retroviral proteins; however, these elements lack the envelope protein that is required to exit the cell. Retrotransposons undergoduplicative transposition, as their total number increases after each transposition with the potential to expand genomes. For example, one non-LTR retrotransposon family, LINE1, constitutes 17% of the total human genome1.
Type II TEs, known as DNA transposons, do not require a reverse-transcription step to integrate into the genome. Instead, a transposon-encoded protein called a transposase recognizes the terminal inverted repeats (TIRs) that flank the TE, excises the TE out of the donor position, and then integrates the transposon into the new acceptor site. The gap that is left at the donor position can then be either repaired without element replacement in cut-and-paste transposition, or filled with a copy of the transposon by gap repair. Helitrons are newly identified DNA transposons that duplicate differently, using a rolling-circle mechanism. Autonomous helitrons contain a DNA helicase protein, as well as a replicase protein similar to replicon protein A (RPA).
TEs can also be classified by their self-sufficiency. Both retrotransposon and DNA-transposon families have autonomous elements and non-autonomous elements. Non-autonomous elements are often mutated relics of autonomous family members, but sometimes have only limited sequence similarity to their autonomous counterparts. Non-autonomous DNA transposons often consist of a pair of TIRs surrounding non-transposon DNA, which is frequently copied from an unrelated protein-coding gene. Additionally, some non-autonomous elements, called miniature inverted-repeat transposable elements (MITES), consist of two TIRs that are connected in a tail-to-tail orientation. The number of non-autonomous elements in a genome can greatly outnumber the autonomous elements. For example, there are an estimated 1.2 million copies of the Alu repeat in the human genome (reviewed in Ref. 122), which is a non-autonomous non-LTR retrotransposon (also known as a short interspersed nuclear element, or SINE).Transposable Elements: http://www.nature.com/
There are 2 types:
Type 1 Copia: Belong to or similar to Pseudovirideae; abundant in algae, bryophytes, gymnosperms and angiosperms.
Type2-Gypsy: it belongs to Metaviridae group they are widely distributed among the plants such as Gymnosperms and angiosperms.
LTR less Retrotransposons:
They are found in large numbers ~ 250 000 copies per plant genomes. Based on structural and functional features they are classified as LINES and SINES.
LINES:
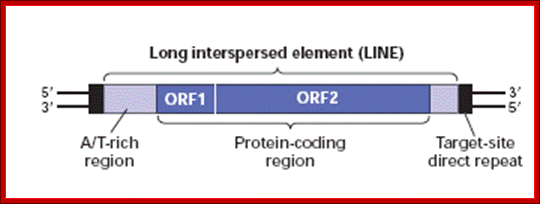
They are 5kb or more in length. They are derived from reverse transcribable RNAs by RNAP II. Lines contain 2 coded segments. One codes for a protein that binds to ssRNA, the other is reverse transcriptase having endonuclease function. They thus enable to copy themselves and transpose. They also do so in the genome of SINES such as Alu elements and transpose.
http://biosiva.50webs.org/
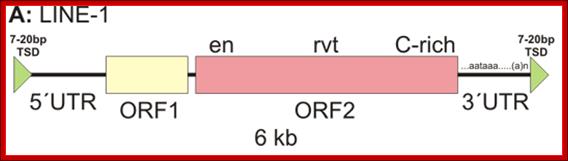
http://biol.lf1.cuni.cz/
A typical LINE consists of 5’UTR and 2 ORFs and a 3’UTR. The 5’UTR contains promoter element for RNAP II and 3’UTR has poly-A additional sequences or signals-AAUAAA.----//--(GU)n. So the transcripts are added with polyA-tail; as it is copied by RNAPII it may be added with 5’cap.
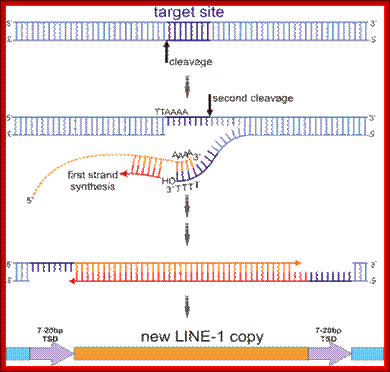
As the elements copy their genome and transpose they contribute to the increase in the genome size. Human genome contains 900,000 copies of such elements which accounts for 21% of the genome.
Lines can be used for genetic finger printing by PCR or RFLP. Retroviruses behave like perfect LINES elements and some Lines are actually derived from them.
SINES:
The size of the SINES’ average length can be 500bp (+/-) 50bp or so). They are reverse transcribed elements but transcribed by RNAP III. The progenitors of SINES are rRNA, tRNA and other small molecular weight RNAs. They don’t encode any RT enzymes perse, but they are transcribed and transposed by Lines components. The most common and abundant Sine element is Alu RNA-280 bp long. They don’t have any coding sequences. They can be recognized and cut by Alu restriction enzyme. The number of SINES is over a million copies account for 13% of the human genome. The most common form of transposons in humans is Alu element, 280bp long, contain Alu site. The number ranges from 300,000 to a million of them.

http://biol.lf1.cuni.cz
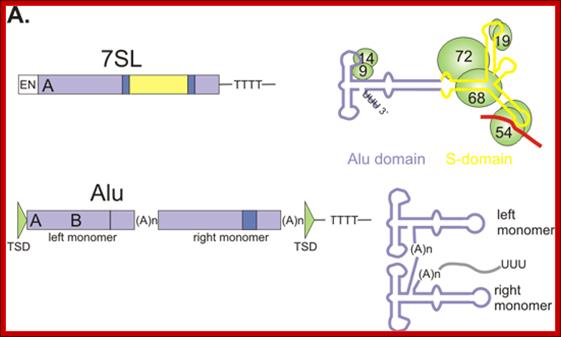
http://www.ncbi,nih,gov
MITES:
Mostly found in plants. They are Miniature Invert Repeat Transposable Elements. They have inverted terminal repeats with 2-3 bp target sequences. No coding sequences, 200 to 500 bp long, found to contain flanking protein coding genes. They are not related to LINES or SINES.
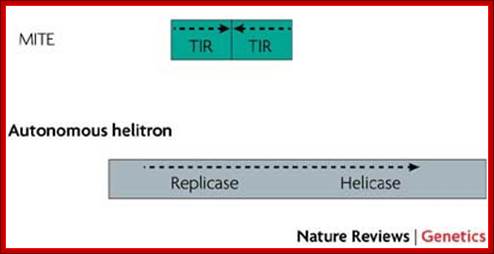
www.nature.reviews
Class II Mobile Elements:
DNA transposons:
Class II mobile elements are also called class II transposons; which are actually DNA transposons. They are more akin to Ac and Ds elements discovered by McClintock.
They were first discovered by Madam Barbara McClintock; she called them as jumping genes. They are causative factors for changing color of endosperm in corns.

https://iweb.langara.bc.ca
Their mode of transposition is totally different from Retrospons. They don’t use RNA as an intermediary. They have the ability to cut its DNA and transpose to other sites by what called cut and paste mode. They use Transposase for transposition.
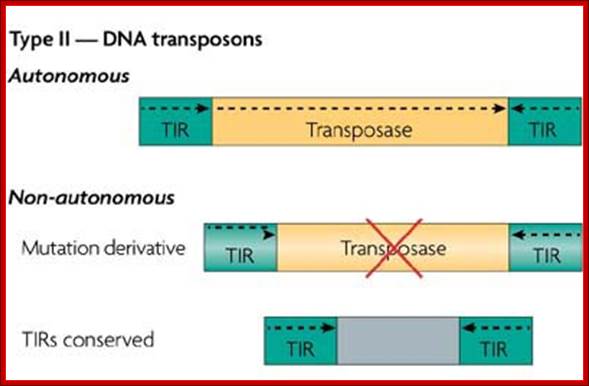
https://www.slideshare.net
There are different types of Type II Transposons and work in different ways. They cut the transposon segment and create sticky ends. They cut the Host DNA anywhere (target) and generate sticky ends. The gap between cut ends of transposon and the target DNA filled up by DNA polymerase and ligation is performed by DNA ligase (seals the ends). The filling reaction creates new regions on either side of the transposons. The flanking sequences can be identified as direct repeats.
Some transposition is site specific. And use certain sequence elements in the target DNA. Not all of DNA transposons perform transposon functions as mentioned above. Some use replicative method of transposition. How, first produce RNA and it is made into ds cDNA and then it is cut and pasted to new target?
Drosophila has acquired another transposable element called P element, nearly 50 years ago, now this is widely spread in Droso population.
In bacteria the DNA transposons called IS elements contain transposase and an additional element that can act against antibiotic. Such transposons are called Tn elements.
Mu phage element is best known for its replicative transposition, similar to homologous recombination coupled with a part of replication.
Transposons and diseases:
Once transposon elements were considered as junk DNA, but now they are implicated in genetic diseases and they have also contributed to the increased genome size and variation and evolution.
Transposons can be considered as mutagens in one way or the other. Insertion in the middle or any part of the gene can disable the function of that gene. If the transposon jumps out of the gene, it can cause problems with repairing.
Multiple copies can bring about unequal crossover and cause mutation at chromosomal level. Introduction of transposable element can destroy the function of a gene, so one can study the effect of gene disruption
Human transposable elements:
Classes of interspersed repeat in human genome:
|
Kinds |
State |
Structure |
length |
Copy |
% |
|
LINES |
auto |
Orf1-orf2pol-AAA |
6.8kb |
8.5x10^5 |
21 |
|
SINES |
Non auto |
-#--A-B—An# |
100-200bp |
1.5x10^6 |
13 |
|
|
Non auto |
#ltr-gag-# |
1.5-3kb |
|
8 |
|
Retro like |
Auto |
#-ltr—gag-pol-envX-Ltr# |
6-11kb |
4.5x105 |
8%
|
|
DNA transposons |
Auto |
#transposase# |
2-3kb |
|
|
|
|
Non-auto |
#-I-I-# |
8—3000bp |
3000 |
3% |
|
|
|
|
|
|
|
Moderately repetitive Genome:
Moderately repetitive DNA such as rRNA, tRNA, 7SL RNA, Sn, sno RNAs and few others vary in their number and types. Many transposable elements have been included in this category.
|
|
Copies x1000 |
Ntds/ in mbs |
Fraction of the genome |
|
|
LINES |
868 |
559 |
20.4 |
|
|
L1 |
516 |
462 |
16.9 |
|
|
L2 |
315 |
88 |
3.2 |
|
|
L3 |
37 |
8 |
0.3 |
|
|
|
|
|
|
|
|
SINES |
1558 |
360 |
13.1 |
|
|
Alu |
1090 |
290 |
10.6 |
|
|
MIR |
393 |
60 |
2.2 |
|
|
MIR3 |
75 |
9 |
0.3 |
|
|
|
|
|
|
|
|
LTR |
|
|
|
|
|
ERV class |
443 |
227 |
8.3 |
|
|
DNA Transposons |
294 |
78 |
2.8 |
|
DNA mediated Transposons:
|
Bacteria replicative transposons |
Terminal inverted repeats, flank- Anti bio resistance & transposase |
Copy DNA and insert the same |
tn3, yd, mu |
|
|
Bacterial cut n paste transposons |
Ter inverted repeats-flank anti- bio resistance n transposase |
Excision of DNA from old and insert at new target |
tn5, Tn10, tn7,Is911,Tn917 |
|
|
|
|
|
|
|
RNA mediated transposons:
|
Viral like retro transposons |
250-600 direct Ter LTRs, Gag n RT ,integrase |
Transcribe from RNA from LTR –reverse transcribe and insert |
Ty elements-yeast Copia in Droso |
|
|
Poly-A retrotransposons |
5”UTR, flank encoding RNA binding proteins and RT 3’A-T rich |
Transcribe into RNA from an internal promoter, target primed by RT initiated an endonuclease |
F n G elements of Droso, Lines and SINE in mammals, Alu in Humans |
|