Glycosylation of Proteins:
Glycoproteins:
Proteins synthesized are modified at specific positions, while they are synthesized Co- translationally and many are modified post translationally. Proteins, thousands of them have their own specific destination and they are delivered to their respective sites; an incredible achievement that happens in every cell. The design and success of this complicated process has taken 3.8 billion years to perfect (not completely) the system and to understand this humans took more than 5000 years, it may take another few more thousand years to understand completely.
Proteins added with glycans (carbohydrates), where protein part is dominant and carbohydrate part is minor- such proteins are called Glycoproteins. On the contrary proteins are added with greater number of chains of carbohydrates, such proteins are called Proteoglycans. Glycoproteins and Proteoglycans are characteristics of eukaryotes, but bacteria too produce glycoproteins in their cytoplasm such as N-linked and O-linked glycosylated proteins. Understanding of bacterial cytoplasmic glycosylation is important in developing proteins for medicinal and research purposes.
Proteoglycans:
Proteoglycans are proteins that are heavily glycosylated. All proteoglycans have a core protein with greater amount of characteristic glycosaminoglycan (GAG) chain(s) attached. The carbohydrate chains are long, linear polymers that are negatively charged under physiological conditions, due to the occurrence of sulfate and uronic acid groups. Proteoglycans occur in the connective tissue, synovial fluids and junctions.
Proteoglycans can be categorized depending upon the nature of their Glycosaminoglycan chains (CH2O groups). These chains may be:
Proteoglycans can also be categorized by size. Examples of large proteoglycans are Aggrecans, the major proteoglycan in cartilage, and Versican, present in many adult tissues including blood vessels and skin. The small leucine-rich repeat proteoglycans (SLRPs) include Decorin, Biglycan, Fibromodulin and Lumican.
Glycosylation of Proteins in Endoplasmic Reticulum:
As soon as the polypeptide chain is pulled or threaded into the lumen, the signal sequences are cleaved by signal peptidases and then they are guided by BiPs/HSPs (Chaperones). It is at this point, proteins are not yet folded to their 3-D conformation, glycosylation and other modifications of proteins start. Such modifications are not a priority, but such modifications help folding in proper context or otherwise such unmodified proteins are susceptible for degradation. Protein modification is just not restricted to ER, but it can take place in Golgi bodies and cytoplasm and even in the nucleus.
Protein modification can and also occurs in nuclear compartment and free cytoplasm. For that matter, only in 1970-1980, the process of nuclear and cytoplasmic glycosylation have been discovered. In fact, during the past two decades, it has become clear that O-GlcNAc is one of the most abundant posttranslational modifications within the nucleocytoplasmic compartments of all metazoans including plants, animals, and the viruses that infect them. Many protein such as nuclear pore proteins, many transcriptional factors, RNA binding proteins, cytoskeletal proteins, chaperones and many regulatory proteins.

(A) O-GlcNAcylated proteins occur in many different cellular compartments. Found mostly within the nucleus, they are also in the cytoplasm and within viruses that infect eukaryotic cells. The subcellular distribution of O-GlcNAc is similar to that of Ser(Thr)-O-phosphorylation. (Redrawn, with permission, from Comer F.I. and Hart G.W. 2000. J. Biol. Chem. 275: 29179–29182.) (B) O-GlcNAcylated proteins belong to many different functional classes. Pie chart illustrates the functional distribution of O-GlcNAcylated proteins identified thus far. (Redrawn, with permission, from Love D.C. and Hanover J.A. 2005. Science STKE 312: re13.), http://www.ncbi.nlm.nih.gov/books
Many proteins are modified internally by modifying single amino acids such as cofactor binding, disulphide bonding, methylation, acetylation, glycosylation, ADP ribosylation, carbamylation, phoshorylation and others in a.a sequence specific manner. Such modifications specify specific structure and function to proteins. The above said modifications are reversible, but there are irreversible modifications such as proteolysis, ubiquitination, peptide tagging, tyrosine hydroxylation and protein splicing.

Chaperone and Chaperonin-mediated protein folding. http://slideplayer.com
(a) Many proteins; They fold into their proper three-dimensional structure with the assistance of Hsp70, a molecular chaperone that transiently binds to a nascent polypeptide as it emerges from a ribosome. Proper folding of some proteins; Many also depends on the chaperonin TCiP, a large barrel-shaped complex of Hsp60 units. (b) GroEL, the bacterial homolog of TCiP, is a barrel-shaped complex of 14 identical 60,000-MW subunits arranged in two stacked rings. In the absence of ATP or presence of ADP, GroEL exists in a “tight” conformational state (left) that binds partially folded or misfolded proteins. Binding of ATP shifts GroEL to a more open, “relaxed” state (right), which releases the folded protein. [Part (b) from A. Roseman et al., 1996, Cell 87:241. Courtesy of Helen Saibil.]
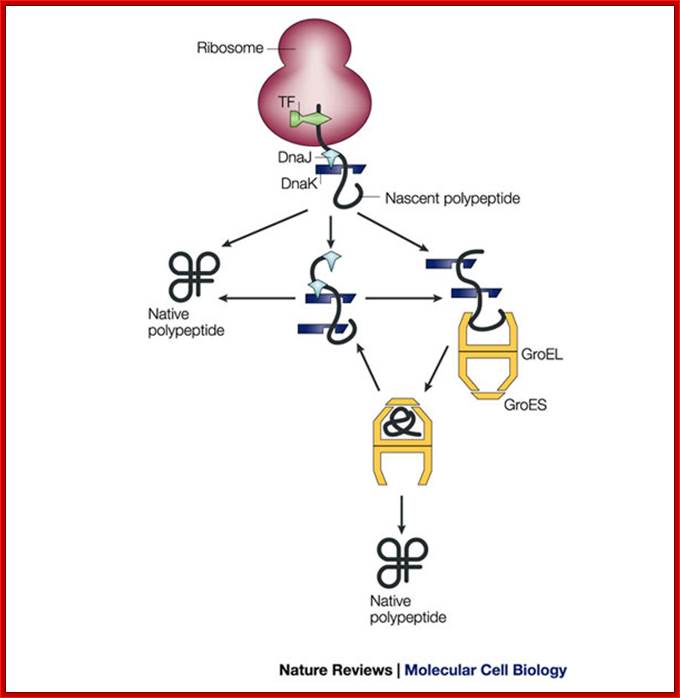
Pathways of chaperone-mediated protein folding in the cytosol; The bacterial chaperonin GroEL consists of two homo-heptameric rings, which form a barrel-shaped structure. Each subunit has an equatorial ATPase domain where the rings contact each other, and an apical substrate-binding domain at the open end of the barrel. Alternating ATP cycles in each ring control the conformations of the apical domains. The GroES co-chaperone is a homo-heptameric structure that can also bind to the GroEL apical domains. After substrate binding to one ring of GroEL (see figure, part a), ATP and GroES are bound by the same ring, which displaces the substrate into an enclosed cavity that is capped by GroES (part b). Sequestration of the substrate (part c) allows folding without the risk of non-productive intermolecular contacts, which could otherwise lead to aggregation. Dissociation of GroES after ATP turnover allows substrate release (part d)19, 20. Some polypeptides require several cycles of binding and release to reach their native state. Certain polypeptides that are too large to fit inside the chaperonin cavity can also be bound by the ring of GroEL that is opposite the ring that is bound by GroES, and their folding can be assisted through cycles of such trans binding of GroES;
Protein modifications:
As proteins are translocated into ER, enzymes/factors found in the lumen assists folding of proteins into 3-D structure and also at the same time proteins are added with certain adducts such as acetyl groups, methyl groups, phosphate groups and others on specific amino acids in sequence specific manner. These modifications have important bearing on their structure, location and function.
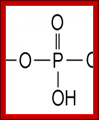
Phosphate group
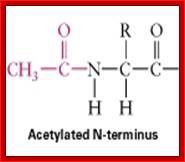

SH group
Organic Functional Groups aredded to side chains of aminoacids in polypeptide chains either in cytosol or ER or both.hydroxyl and methyl groups
- Methyl, -CH2, -CH3, Hydroxyl, ‑OH, Carboxyl, -COOH, –COO, -CHO2, Amino, -NH2
- Sulfhydryl, -SH, Phosphate, -PO4 groups are added to specific aminoacids in P-chain- http://www.nclack.k12.or.us
The internal residues in proteins can be modified by attachment of a variety of chemical groups to their amino acid side chains. The most important modifications are phosphorylation of serine, threonine, and tyrosine residues and methylation of lysine and arginine. There are numerous examples of proteins whose activity is regulated by reversible phosphorylation and methylation. The side chains of asparagine, serine, and threonine are sites for glycosylation. Many secreted proteins and membrane proteins contain glycosylation.
Unlike chemical modification of residues, which often is reversible, processing of some proteins causes irreversible changes that alter their activity. In the most common form of processing, residues are removed from the C- or N-terminus of a polypeptide by cleavage of the peptide bond in a reaction catalyzed by proteases. Proteolytic cleavage is a common mechanism of activation or inactivation, especially of enzymes involved in blood coagulation or digestion. As discussed later, the activity of certain digestive enzymes is controlled by this mechanism. Proteolysis also generates active peptide hormones, such as EGF mentioned earlier and insulin, from larger precursor polypeptides.
Self-splicing, is an unusual type of processing, occurs in bacteria and primitive eukaryotes. Splicing refers to a process analogous to splicing of pre-mRNAs, an internal segment of polypeptide, an intein, is removed and the ends of the extien segments are joined. Unlike proteolytic processing, protein self-splicing is an autocatalytic process, (which again comparable to that of self splicing introns), which proceeds by itself without the involvement of enzymes. In vertebrate cells, processing of some proteins involves self-cleavage, but the subsequent ligation step is absent. One such protein is Hedgehog, which is critical to a number of developmental processes.
For proteins, to fold properly into their 3-D structural form, first they are bound y chaperones, then they are subjected to sulfhydril bond formation at specific positions; the folding is also aided by PDI associated with glutathione, which as an active factor. Chaperones play an important role.
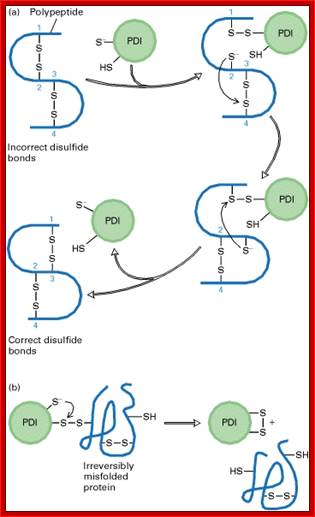
Process of reformation of S-S bonds:
;Protein disulfide isomerase; http://www.studyblue.com/
What is Glutathione - A Master Antioxidant?
Glutathione:
Structure and Creation:
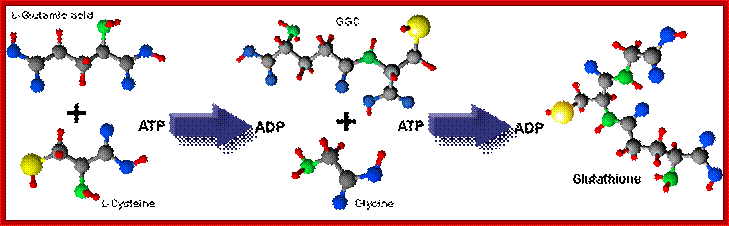
Glutathione is the most biologically abundant (can be more than 5mM per cell) low molecular weight intracellular thiol tripeptide; Glutamine-Cysteine-Glycine. It can dimerize into GSSG. By a way of the reducing power of its free sulfhydryl (-SH), glutathione plays a key role in many cellular processes. These involve protection of cells against oxidative stress, xenobiotics and radiation (reviewed by Sies, 1999 and Sen, 1997). Glutathione is synthesized intracellularly from its constituent amino acids by the consecutive action of two enzymes, γ-glutamylcysteine synthetase (γ-GCS) and glutathione synthetase (GS), with both reactions consuming ATP.
Method of action
Glutathione's main role as an antioxidant is the use of its sulfhydryl group as a nucleophilic scavenger and electron donor. However, glutathione has several other vital maintenance activities.
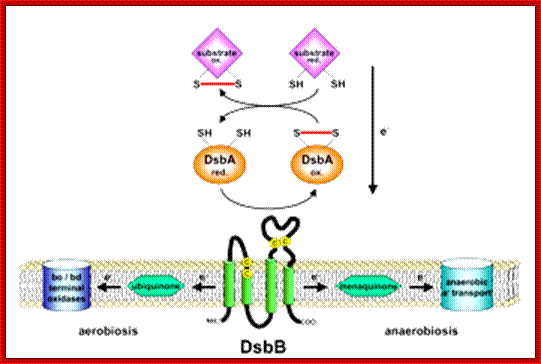
Model of DsbA and DsB system;;Pathway of disulfide bond formation; http://beckwith.med.harvard.edu/
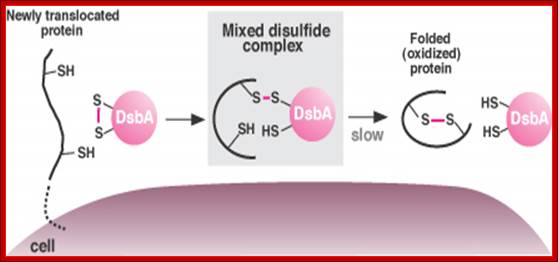
DsbA promotes protein folding in bacteria by donating a disulfide bond (S-S). The process begins when a string of amino acids moves into the pericellular space (left). The protein binds DsbA, exchanging a hydrogen atom for a disulfide bond, which it then uses to fold and stabilize itself (right). Normally, DsbA and the protein dissociate too quickly for the intermediate form, or mixed disulfide bond, to be observed. Certain mutants slow down the process, allowing the mixed disulfide complex to be observed (middle). (Image by Jeff Cleary); http://archives.focus.hms.harvard.edu/
- A cofactor in the function of several antioxidant enzymes, glutathione peroxidises, which are responsible for the degradation of peroxides.
- Maintenance of ascorbate in its reduced state so it can remain an effective antioxidant/reducing agent. It does this through the activity of the glutathione-dependent enzyme dehydroascorbate reductase.
- An essential component in an NADPH pathway that prevents cellular components from being oxidized. This function is essential for the maintenance of protein thiols, and the creation of deoxy ribonucleotide precursors for DNA synthesis, through the reduction of ribonucleotides, (Meister, 1991).

Frederick T. Guilford 1,* and Janette Hope;http://www.mdpi.com
Protein disulphide bond formation and folding:
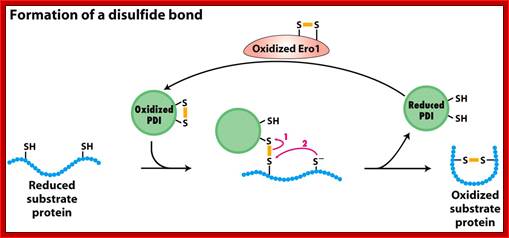
a) In this process the Protein Disulphide Isomerase PDI and tripeptide glutathionine (protein-disulfide reductase) are very essential in ER to produce S-S bond formation. The discovery of ER Oxidation/Rduction components Ero1 as a provider of oxidizing equivalents for the formation of disulphide bonds (Frand & Kaiser, 1998; Pollard et al, 1998; Sevier et al, 2001) led researchers to question the hypothesis that GSSG is solely responsible for the oxidation of PDI in the ER.
b) Proteins have natural tendency for producing disulfide bonds, often these are formed at wrong positions. The protein disulphide isomerase resident of ER lumen contains glutathionine with SH groups. Glutathionine is a tripeptide short protein contains SH groups. PDI contains an active-site with two reduced cysteine sulfhydryl (–SH) groups. The ionized (–S−) form of one of these groups reacts with disulfide (S – S) bonds on nascent or newly completed proteins to form a disulfide-bonded PDI-substrate protein intermediate. This generates a free –S− group on the protein, which, in turn, can react with another disulfide bond in the protein to form a new disulfide bond and another free –S− group. In this way, the disulfide bonds on a protein can rearrange themselves until the most stable conformation for the protein is achieved, and free PDI is released.
(b) An escape hatch for PDI: Occasionally PDI forms a disulfide bond with a protein that is irreversibly misfolded. PDI eventually escapes when the second –S− group in the active site attacks the disulfide bond between PDI and the protein, releasing PDI with an internal (intramolecular) disulfide bond. [Part (a) after R. B. Freedman, 1984, Trends Biochem. Sci. 9:438, and R. Noiva and W. Lennarz, 1992, J. Biol. Chem. 267:3553. Part (b) after K. Walker et al., 1997, J. Biol. Chem. 272:8845.]

The role of glutathione in disulphide bond formation and endoplasmic-reticulum-generated oxidative stress, Seema Chakravarthy et al.
The production of reduced or oxidized glutathione can occur at various stages during the formation of native disulphide bonds. Protein disulphide isomerase (PDI) is used as an example of how the levels of reduced glutathione (GSH) or oxidized glutathione (GSSG) are maintained in the endoplasmic reticulum (ER). (1) Free cysteines are introduced into the ER in the form of newly translocated proteins. (2) Disulphide bonds are formed by PDI, which is reduced. PDI is oxidized by Ero1,which is oxidized by O2, a process that generates reactive oxygen species (ROS). Detoxification of ROS can lead to an increase in GSSG. (3) PDI might also be oxidized by GSSG leading to an increase in GSH. (4) PDI is reduced by GSH, which leads to an increase in GSSG. (5) Influx and efflux of GSH or GSSG from the ER may control their ratio. Cytosolic GSSG is reduced by glutathione reductase. Similar activity might occur in the ER lumen. Chakravarthi et al; http://embor.embopress.org/
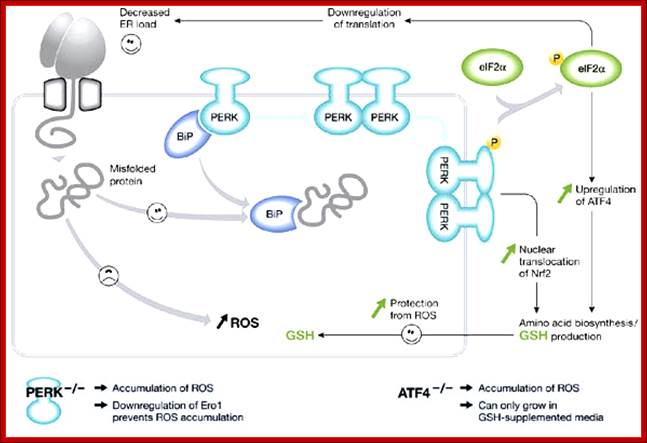
PKR-like ER kinase-dependent stress response to protein misfolding in the endoplasmic reticulum of mammalian cells: The unfolded protein response in mammalian cells leads to the activation of the transmembrane kinases, Ire1α, Ire1β, PKR-like ER kinase (PERK) and the transmembrane activating transcription factor 6 (ATF6). The PERK pathway is activated after dissociation of BiP from PERK monomers, causing dimerization and activation of the cytosolic kinase domain. (Note that the role of BiP in this process is unclear (Credle et al, 2005).) This activation leads to the phosphorylation of eIF2α, causing attenuation of translation and thereby alleviating endoplasmic reticulum (ER) stress. However, translation of transcription factor ATF4 is stimulated and, with activation of NF-E2-related factor 2 (Nrf2), leads to an induction of genes involved in amino-acid synthesis. The cellular level of glutathione is increased, which can potentially protect the cell from damage caused by reactive oxygen species (ROS) produced during ER stress. Perturbation of either the PERK or ATF4 pathway leads to an increase in the production of ER-derived ROS; Seema Chakravarthy etal; http://embor.embopress.org/
A non‐catalytic disulphide bond regulating redox flux in the ER oxidative folding pathway:
The formation of protein disulphide bonds in newly synthesized secretory proteins within the endoplasmic reticulum (ER) depends on an electron transfer system in which oxidizing equivalents are passed from molecular oxygen through several carriers within the oxidase Ero1 to protein disulphide isomerase (PDI) and then to the reduced protein substrate. Recent articles published in The EMBO Journalhighlight a crucial and surprising feature of the control of this process in mammalian cells; the activity of the key oxidase Ero1α is downregulated in oxidizing conditions through the formation of a stable non‐catalytic disulphide by a cysteine residue that participates in one of the redox active sites within the oxidase (Appenzeller‐Herzoget al, 2008; Baker et al, 2008).

(A) In the absence of a reduced substrate, PDI and the FAD and inner disulphide redox centres of Ero1α are oxidised, the regulatory C94–C131 disulphide forms and the glutathione buffer is shifted towards GSSG. (B) In the presence of a reduced substrate, some PDI is converted to the reduced form, which reduces the regulatory disulphide; reducing equivalents flow from substrate protein to O2, all redox centres interconvert between the oxidised and reduced state and the glutathione buffer shifts towards GSH.; Non Catalytic S-S bond formation;Robert Freedman; http://emboj.embopress.org/
Disulfide Bond Isomerase system:
http://gouldinglab.bio.uci.edu/
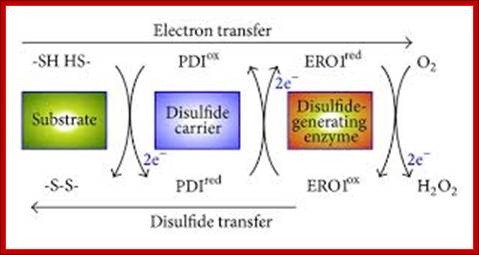
Disulfide relay from ERO1 to PDI in the ER. Disulfide bond formation involves electron transfer from the substrate to a disulfide carrier protein (e.g., PDI) and then to a de novo disulfide-generating enzyme (e.g., ERO1). PDI directly transfers a disulfide to two Cys residues of a substrate protein by means of a thiol-disulfide exchange reaction. The reduced form of PDI is reoxidized by ERO1, which relays the oxidizing power from molecular oxygen, via the FAD cofactor, to the reduced form of PDI.
Disulfide Bond Isomerase system: http://www.hindawi.com/
http://gouldinglab.bio.uci.edu/
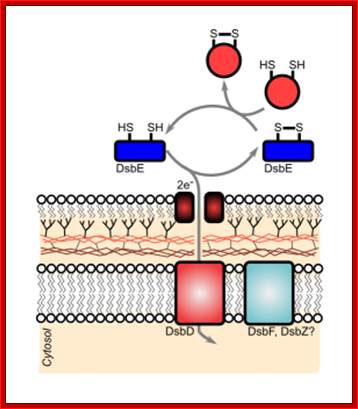
Disulfide bond-forming (Dsb) proteins have been shown to be involved in virulence in many pathogenic bacteria. They are oxidoreductase proteins that have a variety of functions including chaperone activity, electron transfer and disulfide bond isomerase activity. It has been predicted that of the 161 potential secreted proteins of M. tuberculosis approximately 60 % of these contain at least one disulfide bon. Hence, Dsb proteins which assist in folding secreted proteins into their correct conformation and assist in disulfide bond formation are of great importance for the survival of M. tuberculosis. Utilizing bioinformatics, two secreted Dsb proteins have been identified in M. tuberculosis, one of which is my target protein to investigate the secreted disulfide bond isomerase system of mycobacteria. The secreted proteins which these two Dsb proteins interact with is presently under investigation (Goulding Lab). http://gouldinglab.bio.uci.edu/
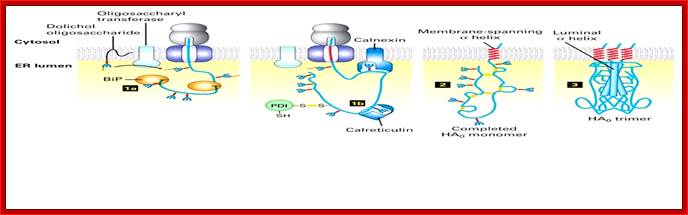
While the nascent chain is still growing, two protein-folding catalysts, calnexin and calreticulin, associate with the protein, and three disulfide bonds form in the globular head domain. Following completion of translation, three additional disulfide bonds form and possibly rearrange in the monomer. Three HA0 chains then interact with each other, initially via their transmembrane α helices; this association apparently triggers the formation of a long stem containing one α helix (dark rod) from the luminal part of each HA0 polypeptide. Finally, interactions between the three globular heads occur, generating the mature trimeric spike. Calnexin and calreticulin bind to N-linked oligosaccharides with a single glucose residue on unfolded protein segments, thereby promoting the proper folding and assembly of newly synthesized glycoproteins such as HA. Most of the properly folded proteins are further modified and transported to their specific destination. [Adapted from sketch by Dan Hebert and Ari Helenius.See M-J. Gething et al., 1986, Cell 46:939; U. Tatu et al., 1995, EMBO J. 14:1340; and D. Hebert et al., 1997, J. Cell. Biol. 139:613.]
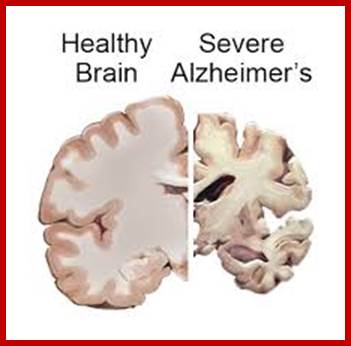
One of the fundamental unsolved problems in biology is how proteins attain their proper three dimensional conformations. Heat shock proteins and protein folding catalysts are vital in assisting proteins in this process. It is interesting in understanding the molecular mechanism of these folding helpers. Alzheimer's, disease is the result of defective folding and unfolding and aggregating.
Alzheimer’s and Parkinson’s disease:
On their own, Alzheimer’s disease and Parkinson’s disease cause the slow regression of mental and physical abilities. But when the proteins behind these neurodegenerative disorders join forces, a dramatic decline in cognition often results, researchers with UC Irvine’s Institute for Memory Impairments and Neurological Disorders have found. Little has been known about how these disease-inducing proteins interact and influence mental processes, but a group headed by Frank LaFerla, Chancellor’s Professor of neurobiology & behavior and UCI MIND director, work is beginning to change that.
Neurological researchers have found a new therapeutic target that can potentially lead to a new way to prevent the progression of Alzheimer’s disease. The target called neutral sphingomyelinase (N-SMase) is a protein that when activated, can cause a chain of reactions in the cell leading to neuronal death and memory loss.
“There are multiple, neurotoxic, disease-causing pathways that converge on the neutral sphingomyelinase that can cause neuronal loss in the brain of an Alzheimer’s patient,” said, a lead investigator Kalipada Pahan, “If we can stop the activation of the neutral sphingomyelinase, we may be able to stop memory loss and the progression of Alzheimer’s disease.”
Adrian; http://www.elements4health.com
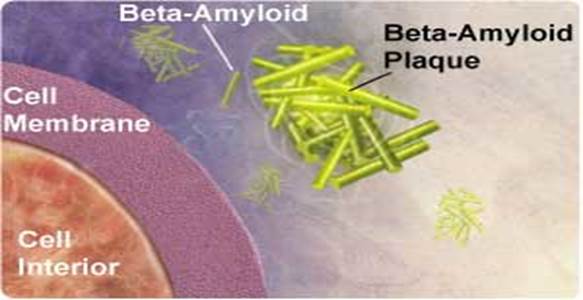
One of the hallmarks of Alzheimer’s disease is the deposition of amyloid beta protein in clumps or “plaques” within the brain. These plaques can be measured in humans with PET scans that use a chemical marker or radiotracer called 11C-PIB.
The prion protein, notorious for causing fatal neurodegenerative disorders such as Creutzfeldt-Jakob disease and mad cow disease, may also be an accomplice in Alzheimer's disease, it's not an infectious misfolded prion protein causing the problem but the cellular form, whose function is relatively unknown.

Showing Amyloid plaques plaque; Biochemistry for medicos-http://www.namrata.co/
Alzheimer's disease is marked by clumps of protein fibers called amyloids that accumulate into "plaques" around nerve cells in the brain, leading to the progressive loss of function of cells. The main protein fragment found in these plaques, amyloid-ß peptide, is created by the improper cleavage of a protein called amyloid precursor protein (APP). Over time, amyloid-ß peptides transform into small clusters known as oligomers, which then develop into the long, sticky fibers that form plaques around brain cells. Scientists are uncertain if amyloid-ß oligomers act directly or through cell surface receptors to affect thinking and decision-making, but most agree that they're toxic to brain.
Bapineuzumab Might Tackle Alzheimer’s Protein Early On?
By Shirley S. Wang
Alzheimer’s-Amyloid clumps; https://www.nia.nih.gov


The hard part is that most people can’t cut carbohydrates out of the body altogether, and in many respects this can be unhealthy. So another way to produce ketones is by consuming oils made from medium-chain triglycerides. MCT is an oil that is made from coconut and palm oil. Mr. Newport began to take coconut oil twice a day at a point where he could not even remember how to draw a clock.
http://humansarefree.com;http://www.dailymail.co.uk
Many scientists believe that Alzheimer’s disease is caused in part by a sticky substance called amyloid that clumps in the brain to cause plaques. But scientists also have posited that the amyloid protein could be causing trouble even before it forms plaques, when it is still floating around in cerebrospinal fluid in its so-called soluble form. (Other theories suggest that other proteins, such as tau, also play a role.)
The experimental Alzheimer’s vaccine Bapineuzumab, being co-developed by Pfizer and Johnson & Johnson, appears to bind to soluble amyloid and “neutralize” amyloid’s downstream toxic effects, preventing it from binding to neurons, according to research presented today at an Alzheimer’s and Parkinson’s disease conference in Barcelona.
Parkinson's disease: (Wikipedia). It is (also known as Parkinson's, idiopathic Parkinsonism, primary Parkinsonism, PD, or paralysis agitans), a degenerative disorder of the central nervous system. The motor symptoms of Parkinson's disease result from the death of dopamine-generating cells in the substantia nigra, a region of the midbrain; the cause of cell death is unknown.

https://www.macalester.edu
The pathology of the disease is characterized by the accumulation of a protein called alpha-synuclein into inclusions called Lewy bodies in neurons, and from insufficient formation and activity of dopamine produced in certain neurons within parts of the midbrain. Mutations in specific genes have been conclusively shown to cause PD. These genes code for alpha-synuclein (SNCA), parkin (PRKN), leucine-rich repeat kinase 2 (LRRK2 or dardarin), PTEN-induced putative kinase 1 (PINK1), DJ-1 and ATP13A2.
Post-Translational Modifications and Quality Control in the Rough ER:
Newly synthesized polypeptides in the membrane and lumen of the ER undergo five principal modifications before they reach their final destinations:
1. Formation of disulfide bonds 2.Proper folding, 3.Addition and processing of carbohydrates, 4. Specific proteolytic cleavages and 5. Assembly into multimeric proteins.
The first two and the last of these modifications, which take place exclusively in the rough ER, are discussed in this section. Although addition of some carbohydrates and some proteolytic cleavages also occur in this organelle, many more such modifications take place in the Golgi complex or forming secretory vesicles; we discuss these in later sections.
Only properly folded and assembled proteins are transported from the rough ER to the Golgi complex and ultimately to the cell surface or other final destinations. Unfolded, misfolded, or partly folded and assembled proteins are selectively retained in the rough or smooth ER, or they are retrieved from the cis-Golgi network and returned to the ER. Misfolded proteins and unassembled subunits of multimeric proteins often move from the ER lumen back into cytoplasm through the translocon where they are degraded in Proteosomes. This pathway is called ERAD pathway. Consider several examples of such “quality control” in the second half of this section.
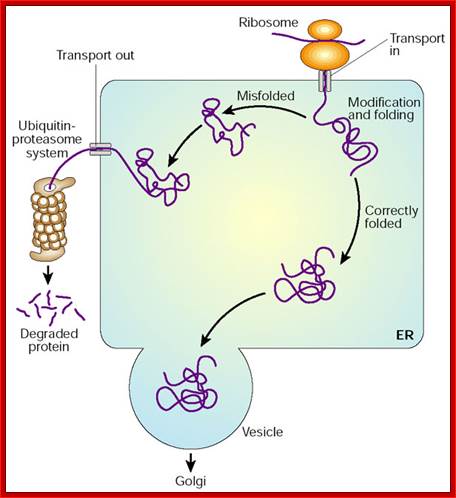
Misfolded and unfolded proteins are translocated out of ER into cytoplasm where they are degraded by proteasome; personal.us.s/vgoder/research
What happens to unfolded-proteins? Unfolded protein Response (UPR):
This is fascinating idiom- “from Cradle to Grave”. This applies to all proteins in all living beings. Unfolded proteins, especially in the lumen of ER, can create problems for the cell. Unfolded proteins don’t move forward and clog the lumen for incoming proteins to move forward. So cells have designed a way by which such proteins are transported out by what is called retrograde pathway and the same is fed into proteasome for degradation. A receptor IRE1 (Inositol Requiring Element or protein) is a transmembrane protein in the inner nuclear membrane, and the outer membrane is continuous with the ER membrane. This multifunctional protein (green) has a binding site for unfolded proteins (blue) on its luminal surface; its nuclear-facing domain contains a protein kinase of unknown function and a specific RNA endonuclease.
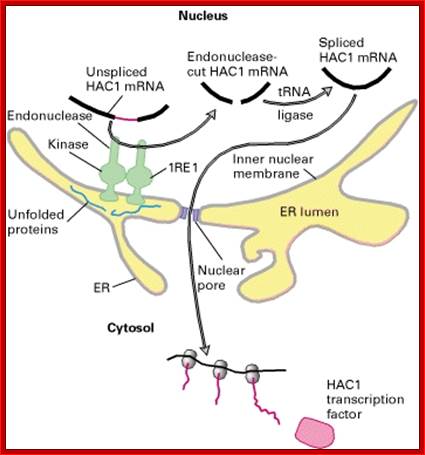
Pre Events for ERAD Pathway; epidemiologiamolecular.com
Binding of unfolded proteins in the ER lumen dimerizes the receptor and somehow activates the endonuclease at the nuclear surface, which cleaves the unspliced mRNA precursor encoding the transcription factor homologs of APAF and CED4 Core apoptotic executioner protein and (HAC1). The two exons of HAC1 (Homologous to Atf/Creb1-bZIP protein) mRNA then are linked together by tRNA ligase, which usually splices tRNA precursors; forming a functional HAC1 mRNA; which moves out of the nucleus. Following its translation, the protein HAC1 moves back into the nucleus and activates transcription of genes encoding several chaperones and other proteins that assist in folding unfolded proteins in the ER lumen. [See C. Sidrauski and P. Walter, 1997, Cell 90:1031.]
Quality control in the endoplasmic reticulum: retrograde protein transport and its end in the Proteasome:
The endoplasmic reticulum (ER) is a membranous organelle present in all eukaryotic cells, designed to deliver proteins to the plasma membrane, the lysosome (vacuole) or to the extracellular space. Such secretory proteins are synthesized in the cytoplasm and enter the ER in an unfolded form through a channel in the ER membrane. After entry into the ER proteins are folded, assembled and packed to be sent from there to their locus of action. Malfolded and therefore inactive molecules which otherwise would block the loci for active molecules have to be sorted out and destroyed. Thus the ER must contain a proofreading system which detects malfolded protein molecules, sorts them out and degrades them.
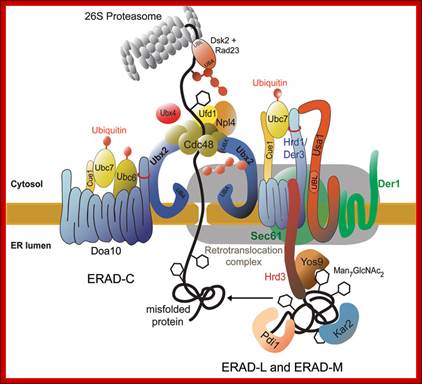
Ubiquitylation in the ERAD pathway; Retrograde transportation; https://www.landesbioscience.com; http://www.cell.com
Up till now it was a biological dogma that proteins that had entered the ER via the import channel were trapped in the organelle and destined for further secretion. It was therefore completely unexpected to discover, that after having detected that a protein is malfolded, this protein was retrograde transported from the ER back into the cytoplasm. Studies on a mutated carboxypeptidase yscY or a mutated ABC transporter Pdr5 showed that retrograde transport is followed by ubiquitination and degradation by the proteasome. These findings in yeast lead to an understanding of some diseases and processes that lead to disease. The hereditary disease cystic fibrosis is linked to degradation of a mutated cystic fibrosis transmembrane conductance regulator (CFTR) molecule which, like Pdr5, belongs to the ABC transporter family. Because of the mutation CFTR never reaches its locus of action, the plasma membrane, but is retained in the ER membrane instead where it is degraded via the ubiquitin-proteasome system. Viruses like cytomegalovirus or the human immunodeficiency virus type I (HIV type I) use the mechanism of retrograde protein transport from the ER to the cytoplasm and the proteasome to get rid of immunoprotective proteins of the host. Also toxins reach the cytoplasm of the host via the same retrograde transport system, where they develop their in part deadly effects. One can expect that studies on yeast will lead to further discovery of target molecules of this process which may be modulated by drugs to avoid development of disease. A protein complex the Cdc48-Udfd1-Npl4 is involved in retro translocation from ER to cytosol where it is subjected to ubiquitination and proteasome mediated degradation.

Molecular Biology of the Cell, Chapter 12 Part4.
If proper folding and assembly fail to occur, misfolded proteins may be removed by cellular quality control mechanisms. The ER-Associated Degradation (ERAD) diagrammed here involves translocation to the cytosol (retrotranslocation, through a modified translocation complex, associated with exit proteins), followed by ubiquitination and proteasomal degradation in the cytosol.
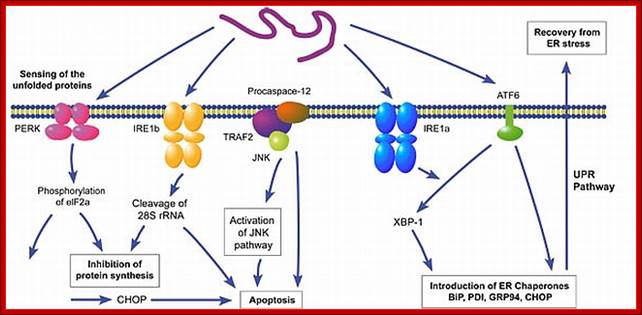

http://bsw3.naist.jp
The ER participates in cellular quality control and in maintaining physiologic
homeostasis. When proteins accumulate in the ER (particularly when they are
misfolded), leads to ER stress. One compensatory mechanism is the unfolded
protein response (UPR), by which production of chaperones is upregulated
through the activation of a unique signaling ER sensor and signal transduction
mechanism (to "handle" the excess unfolded protein). Under severe ER
stress, an ER overload response (EOR), also involving ER signaling,
may trigger upregulation of cytokines, apoptosis or cell death. If interested,
these mechanisms are discussed more fully in Albert et al., Molecular Biology
of the Cell (and only a short notes type in Essentials of Cell Biology).
Three Major Responses to ER stress:
(1) ERAD : ER Associated Degradation, Misfolded proteins are removed from the
ER into the cytoplasm where they are degraded by the ubiquitin proteasome
system.
(2) UPR : Unfolded Protein Response; this is the pathway to decrease misfolded
proteins in the ER lumen. This pathway activates the regulation of protein
translation and the production of chaperone proteins which assist protein
folding.
(3) Induction of Apoptosis; If ER stress continued for a certain period,
programmed cell death is triggered. This response has close relation to
neurodegenerative disease or cancer. (1) ERAD or (2) UPR can be regarded as a
mean which prevents from adopting this worst scenario.
Enteric infection meets intestinal function: how bacterial pathogens cause diarrhea: Retrograde trafficking of cholera toxin. K. Viswanathan, Kim Hodges & Gail Hecht.
After entering the cell, cholera toxin (CT) is routed in a retrograde manner through the Golgi apparatus into the endoplasmic reticulum (ER). A specific amino acid sequence, KDEL, which is located within the A2 subunit of the toxin, mirrors an epitope that is present in proteins that are typically retained in the ER and results in CT translocation from the Golgi to the ER by a shuttle protein known as ERD2. Cholera toxin co-opts the ER-associated degradation (ERAD) pathway to subsequently gain entry into the host cell cytosol. The ERAD pathway ensures that proteins transiting through the ER for secretion are properly folded. Cholera toxin mimics a misfolded protein and is retrotranslocated into the cytosol, where typical ERAD targets would be degraded by the proteasome. Instead, the A1peptide of cholera toxin goes on to ADP-ribosylate adenylate cyclase, leading to production of cyclic AMP (cAMP), which activates protein kinase A. Protein kinase A (PKA) then phosphorylates cystic fibrosis transmembrane regulator (CFTR), leading to Cl− secretion.
Cells possess several quality control mechanisms for a proper folding and function of protein. Malfunction of the mechanisms causes some protein folding diseases. Many proteins are modified with a glycosyl phosphatidyl inositol (GPI) anchor in the endoplasmic reticulum (ER), but the quality control mechanisms of GPI-anchored proteins are not clear, so far. We developed a model misfolded GPI-anchored protein (Gas1*p). Gas1*p can be modified with a GPI anchor in ER, however, the modified Gas1*p was excreted and degraded rapidly via a proteasome. We found that deacylation of GPI by an enzyme (Bst1p) plays important role in the quality control of GPI-anchored proteins.

Figure: Proteins are monitored by quality control systems that ensure correct folding before exiting from the ER. There are a number of molecular chaperones and enzymes to assist proper protein folding in the ER. Misfolded proteins that fail to pass the quality control checkpoint are transported back to the cytosol, and degraded by the Proteasome. GPI inositol deacylation by Bst1p is required for the quality control of GPI-anchored proteins. http://www.keywordsking.com
If proper folding and assembly fail to occur, misfolded proteins may be removed by cellular quality control mechanisms. The ER-Associated Degradation (ERAD) here involves translocation to the cytosol (retro-translocation, through a modified translocation complex, associated with exit proteins), followed by ubiquitination and proteasome degradation in the cytosol.
The ER participates in cellular quality control and in maintaining physiologic
homeostasis. When proteins accumulate in the ER (particularly when they are misfolded),
conditions of ER stress is often created. One compensatory mechanism is
the unfolded protein response (UPR), by which production of chaperones
is upregulated through the activation of a unique signaling ER sensor and
signal transduction mechanism (to "handle" the excess unfolded
protein). Under severe ER stress, an ER overload response (EOR), also
involving ER signaling, may trigger upregulation of cytokines, apoptosis
or cell death. These mechanisms are discussed more fully in Albert et al., Molecular
Biology of the Cell (and only a little in Essentials of Cell Biology).
Glycosylation of proteins in ER:
In general most of the proteins that pass through ER get glycosylated. Glycosylation is site specific, protein specific and tissue specific; for the same protein, glycosylation is different in different tissues. There are four types of glycosylation’s; 1. N-linked:, 2.O-linked, 3. C-end linked or GPI linked and 4. Glycation.
Glycosylation and Cancer:
Almost 50% of all known proteins and more than 80% of
membrane proteins are glycosylated, yet, till recently carbohydrates were
believed be without any functions. Research in the last two decades has shown
that the carbohydrate modifications on proteins and lipids are the key factors
in modulating their structure and function. Only the proteins destined to be on
the membranes or meant to be secreted out were believed to be glycosylated, as
the major glycosylation machinery exists in endoplasmic reticulum and Golgi.
Recent developments however, show that even nuclear and cytoplasmic proteins
are glycosylated.
Cancer cells show altered glycosylation on the cell surface as well as nuclear
and cytoplasmic proteins, Rajiv Kalraiya.
· Many proteins are at first glycosylated in ER and then they are transported into Golgi membranes; there they may be further modified in different ways in different tissues. Glycosylation and sulfide bond formation helps in proper folding. Glycosylation preempt folding. It also helps in multimerization of proteins, ex. Hemagglutinin protein.
· There are specific enzymes for glycosylation in both ER and Golgi complex.
· In general glycosylation precursor builds up at cytosolic surface of the ER from monosaccharides to disaccharides and then to oligo-saccharide clusters. Such glycosylated clusters are transferred to luminal side; then they can be further modified, and added to proteins in sequence specific manner. Even within the lumen one or more glycans are added or removed; depends upon the protein and its destination. The most intriguing aspect is what or which protein to be glycosylated?
· The most common amino acids in the protein to which the carbohydrate oligos are added are Asparagine, and Serine and Glycosyl Phosphatidyl Inositol GPI is added adduct; such glycosylation is called Asn-linked or N-linked glycosylation, serine O-linked glycosylation and GPI linked glycosylation (Glypication) respectively.

Various types of glycosidic linkages: http://www.piercenet.com/browse.cfm?
Glycosylation is a critical function of the biosynthetic-secretory pathway in the endoplasmic reticulum (ER) and Golgi apparatus. Approximately half of all proteins typically expressed in a cell undergo this modification, which entails the covalent addition of sugar moieties to specific amino acids. Most soluble and membrane-bound proteins expressed in the endoplasmic reticulum are glycosylated to some extent, including secreted proteins, surface receptors and ligands and organelle-resident proteins. Additionally, some proteins that are trafficked from the Golgi to the cytoplasm are also glycosylated. Lipids and proteoglycans can also be glycosylated, significantly increasing the number of substrates for this type of modification.
Glycosylation, the attachment of sugar moieties to proteins, is a post-translational modification (PTM) that provides greater proteomic diversity than other PTMs. Glycosylation is critical for a wide range of biological processes, including cell attachment to the extracellular matrix and protein-ligand interactions in the cell. This PTM is characterized by various glycosidic linkages, including N-, O- and C-linked glycosylation, glypiation (GPI anchor attachment) and phosphoglycosylation. Glycoproteins can be detected, purified and analyzed by different strategies, including glycan staining and visualization, glycan crosslinking to agarose or magnetic resin for labeling or purification, or proteomic analysis by mass spectrometry, respectively; http://www.piercenet.com/
|
Types of Glycosylation. |
|||
|
Glycan bind to the amino group of asparagine in the ER |
|||
|
O-linked O-linked |
Monosaccharides bind to the hydroxyl group of serine or threonine in the ER, Golgi, cystosol and nucleus |
|||
|
Glycan core links a phospholipid and a protein |
|||
|
Mannose binds to the indole ring of tryptophan |
|||
|
Glycan binds to serine via phosphodiester bond |
|||
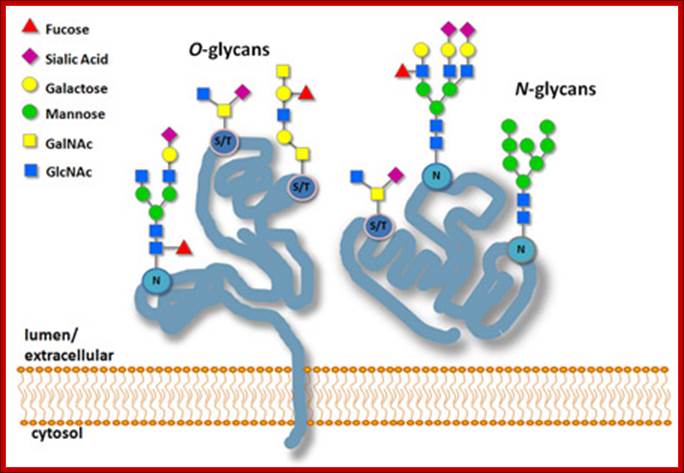
N and O linked Glycation -Glycobiology-New England-Biolabsn
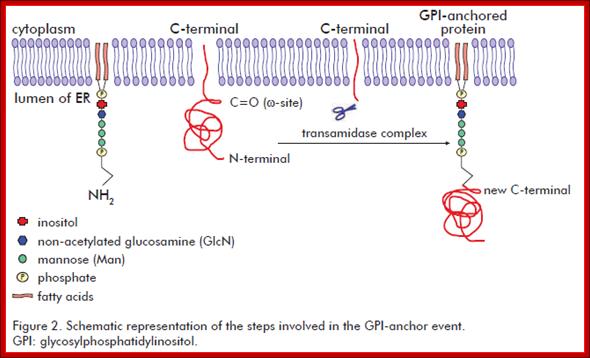
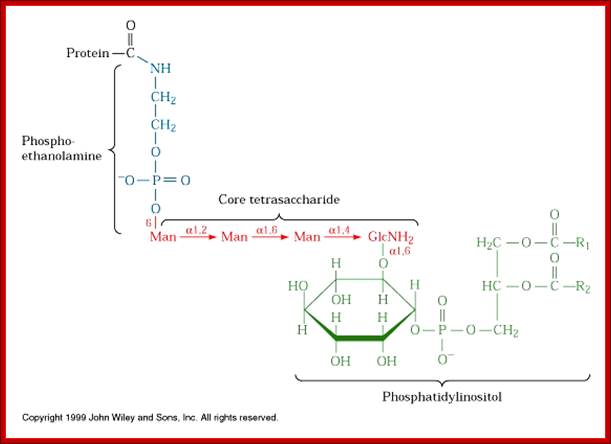
It involves the addition of a GPI molecule to the C-terminal end of the target protein. The GPI molecule is composed by a phosphatidylinositol group and a sugar moiety. The sugar moiety comprises a non-acetylated glucosamine attached to three mannosyl residues and to the phosphatidylinositol group. A phosphoethanolamine residue connected to the terminal mannose mediates the binding of the GPI to the C-terminal end of the mature protein by an amide linkage. Biotecnol Apl v.28 n.1 La Habana ene.-mar. 2011
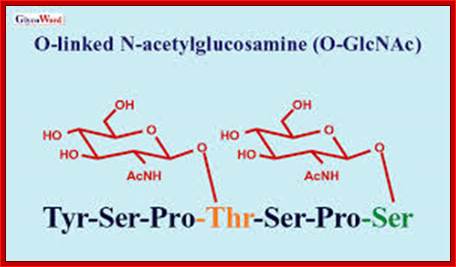
???
· This N-linked glycosylation is more or less restricted to ER, when the protein moves into Golgi complex it may undergo further modifications. The O-linked and GPI linked glycosylation take place in G. complex (Golgi complex).
· In Golgi complex, O-linked glycosylation, where sugars are added to the OH group of serine or threonine. In some cases the C-terminal regions of proteins are also covalently bonded to the amino group ethanolamine of GPI, which is the resident of membranes.
N-linked Glycosylation:
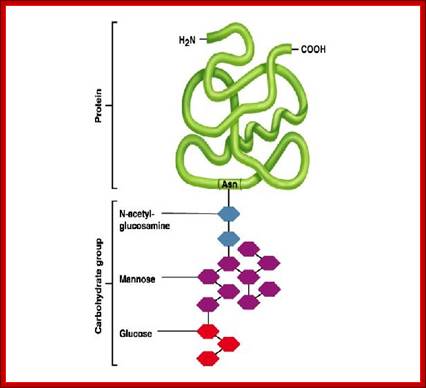
A group of monosaccharides are added in ER and then they are trimmed as they pass on to Golgi sacs. These glycans generally consists of NAG (N-acetyl Glucose amine), mannoses and glycans (14 of them).
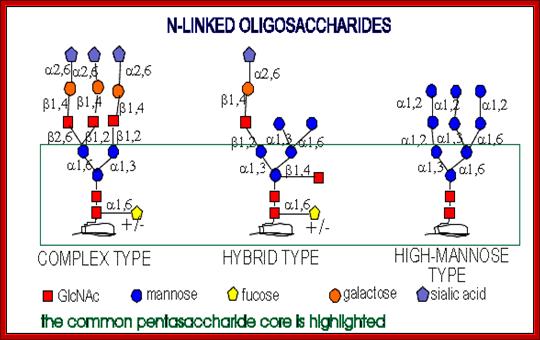
All of them have pentasaccharide core and they are synthesized from common precursor oligosaccharides. The N-linke oligosaccharides have a minimum 5 sugar residues. http://www.cryst.bbk.ac.uk/
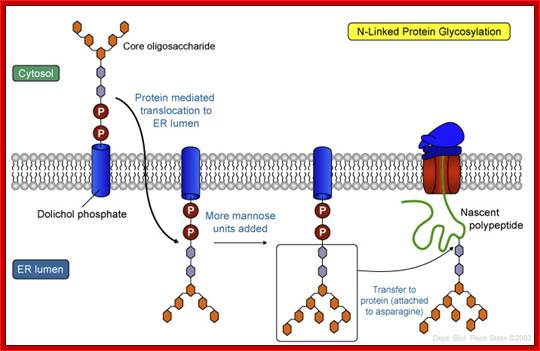
https://www.pinterest.com
The mechanism of N-linked protein glycosylation. The core oligosaccharide is assembled and attached to dolichol phosphate (a lipid carrier) on the outer ER membrane. This lipid-oligosaccharide complex is flipped across the lipid bilayer (so it is now facing the ER lumen) by proteins referred to as flippases. Then, the oligosaccharide is elongated by specific enzymes. Finally, it is transferred to the nascent polypeptide while it is being translated. In N-linked glycosylation, the addition of an oligosaccharide to the polypeptide occurs by a covalent attachment of the sugars to an asparagine side chain; https://wikispaces.psu.edu
Complex type chains can be mono-, bi-, tri- (2,4 and 2,6 branched), tetra-, and pentaantennary structures ( big variety). They can also contain different amounts of sialic acid. High-mannose oligosaccharides can have from 3 mannose residues to as many as 60 in protozoans and yeast.
N-linked glycoproteins exhibit many glycoforms. Cells synthesize many variants of a given glycoprotein. Each variant (glycoform) differs somewhat in the sequences, locations and numbers of its covalently bound oligosaccharides, Example N-linked oligosaccharide chain of RNase B is microheterogenous : a sixth mannose residue occurs at various positions on the core (GlcNAc)2 (Man)5.
RNase B
The diagram shows different types and pattern of carbohydrate complexes are added to proteins in ER; all are N-linked. http://www.cryst.bbk.ac.uk/
Glycosylation in ER is mostly mediated by Dolichol phosphate; it is a polymer of isoprene units; it acts as a carrier of oligosaccharides. Dolichol phosphate is an isoprenoid compound (90-100 carbons total) made from dolichol (synthesis of dolichol via the cholesterol biosynthetic route) by phosphorylation catalyzed by a kinase that uses CTP as the energy and phosphate source. Dolichol phosphate performs two important functions in synthesis of N-linked glycosylation of proteins. It acts as a carrier of Glyco groups. Dolichol is a multimer of isoprene (pentameric carbon skeleton) units, the number of units vary from 20 to 52. The terminal CH2OH is added with phosphate.
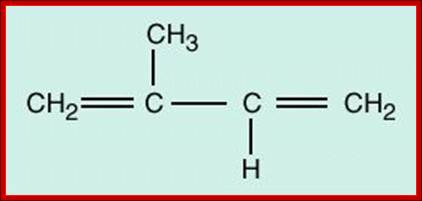
One isoprene unit

Poly-isoprene units; http://www1.biologie.uni-hamburg.de

Dolichol phospho-mannose (Dol-P-Man) is an isoprenoid compound (90-100 carbons total) made from dolichol phosphate. It carriers mannose to the site of assembly of oligosaccharides to be used to make N-linked glycoproteins. Dol-P-Man is made from dolichol phosphate and GDP-Mannose.Dolichol-P mannose-polymer consists of 14-24 (52 in mammalian cells) isoprene units; https://web.squ.edu.om/
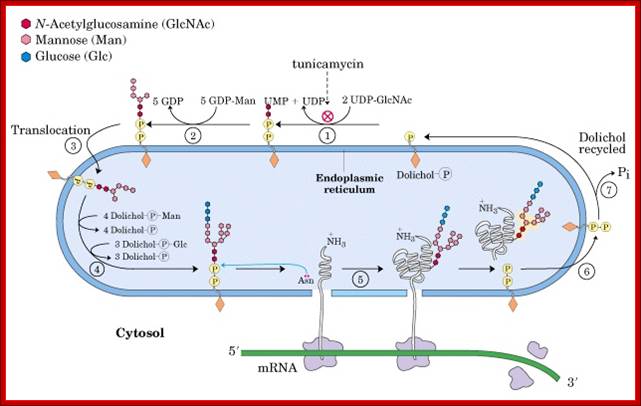
Dolichol phosphate is the isoprenoidal structure upon which the complex oligosaccharides are built before transfer to the target protein. The addition of the first moiety, UDP-N-acetyl glucosamine, can be blocked by the antibiotic, Tunicamycin.
The carrier molecule is Dolichol phosphate, which is anchored to lipid, and it is the resident of ER membranes. It is made up of several isoprene units from 14 to 52 (called polyprenoid). The number of units may vary from 14 to 21 in animal systems, 14 to 52 in plants and fungi and other systems. In these, the alpha isoprene unit is saturated; it is to which phosphate groups are added to generate Dolichol pyrophosphate.
[H2C=C-CH3-CH=CH2] n-H2C=C-CH3-CH=CH2-O~P-O~P

https://en.wikibooks.org
· To begin with oligo-saccharides build up on a carrier molecule at cytosolic side of the ER. Then it is transferred towards luminal side of the ER. Then the oligo saccharides are transferred to proteins; Glycosylation help proteins to fold properly.
· The N-oligo transferases are made up of three subunits. Two of them are ribophorins; integral parts of ER found facing cytosolic surface. The third one is localized in the lumen of ER; it is this subunit that has transferase activity.
After assembly of the oligosaccharides is complete on cytosolic side of ER, the Dolichol-P~P –CH2On structure is flipped by flippases onto ER luminal side.
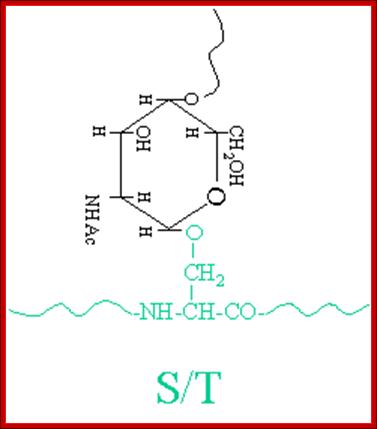
Then the oligo group is transferred from Dolichol phosphate to an asparagine residue of a target protein having the sequence Asn-x-Ser/Thr, where X is any amino acid. Interestingly such basic oligos transferred from Dolichol-P~O~P- are further added with one or more Glycans and this site is domain specific. By the time oligo’s are transferred protein chain is ready to be folded into 3-D state.
1. Dolichol phosphate is a carrier of sugars for oligosaccharide chain synthesis and assembly. Such activated sugars include dolichol-P-mannose and dolichol-P-glucose.
2. Note, Flippases are transmembrane proteins perform transfer across the membranes. Flippases are energy dependent. Some perform without the aid of ATP to perform Flip-Flop functions.
· Glycosylation of a single protein can be at different sites. The kind of glycosylation and position of glycosylation depends upon the conformation of the local sequence.
· The same type of polypeptides in different tissues can be glycosylated in different ways. Ex. Heme-agglutinin glycoprotein of influenza decorated with one type of O-liked glycosylation in one tissue, and if the virus infects different tissue the O-linked decoration for the same protein is different in position and kind.
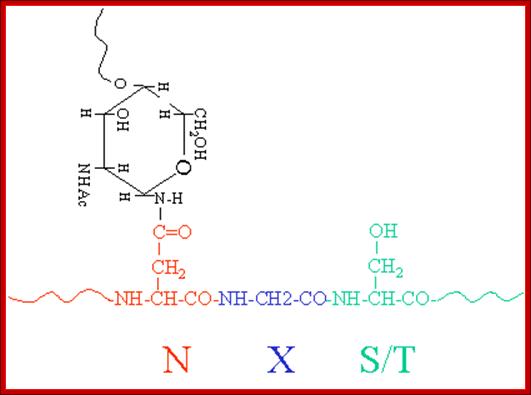
Repeat, step by step:
· Dolichol phosphate located in ER membrane found facing cytoplasm acts as the substrate for the construction of oligosaccharide cluster or tree.
Dolichol + 2CTP ----> Dolichol-P~ o ~P + 2CDP,
· Two successive Glycosyl transferase reactions add two (N-acetyl glucosamine (NAGs) to Dolichol-P-O-P to produce Dolichol-P-o-P-NAG-NAG. Here UDPs act as carriers
Dolichol-P-O-P + 2UDP-NAGs-----> Dolichol-P-O-P-NAG- NAG + 2 UDPs.
· Then 5 mannoses are individually added successively by mannosyl transferases, resulting in a branched mannose oligos. This takes place at cytosolic surface.
Dolichos-P-o-P-+5GDP-mannose ---> Dolichol–P-O-P- (NAG)2-Man (5),
· At this stage, Dolichol-NAG(2)-Man (5) cluster flips from cytosolic surface towards ER luminal side by a translocator protein or flippase, which are specific.
· In the luminal side 4 more mannose residues are added in individual steps to the existing mannose residues,
Dolichol-pp-NAG (2)-Man (5) + 4 Mannose ---> Dolichol-pp-NAG (2)-Man (9),
· Here three branches of mannose are produced. Then another 3 Glycosyl residues are added to one of the three branches of Mannose chains. This results in the formation of Dolichos-p~p-2NAG-9Man-3glu.
· This cluster as a group is transferred on to the target protein at aspargine located at specific motif with specific sequence. Though there are many aspargine residues found in the protein only those aspargine located in a specific motif and with a specific sequence Aspargine-serine-X (the X can be any amino acid residue except proline) selected for transfer. Tunicamycin inhibits the transferase enzyme activity, thus prevents glycoprotein formation.
· Mannoses or glucose or any other monosaccharides at the luminal side are provided as individual components in the form of Dolichol-phosphate-mannose or glucose complex.
· The first cluster of 2 NAG and 5 Mannose formed at cytosolic surface on Dolichol-phosphate backbone as substrate. Then they are internalized. It is from the internalized Dolichol-p-mannose, more mannose is transferred onto the already existing oligo cluster; still linked to Dolichol-pp-oligo saccharide clusters. Specific enzymes perform each of the steps.
Dolichol-o p + GDP-Mannose -> Dolichol-o-p-Man + GDP,
Dolichol-o p + UDP-glucose àDolichol-o P-Glucose + UDP,
· Similarly, individual glycans added on to Dolichol-p~p substrate to produce Dolichol-p~p-glycans at cytosolic side; then they are flipped towards luminal side. It is from this, the glycans are added to the Dolichol-p~p-oligo clusters (2 NAG + 5 Mannose). The fully formed oligosaccharide cluster of 14 sugars is developed inside the ER. However before they are transferred into Golgi complex, few glucoses and few mannoses are removed from the cluster to generate high mannose oligo cluster. In Golgi complex, at different positions, some residues are removed and some are added. Some are modified.
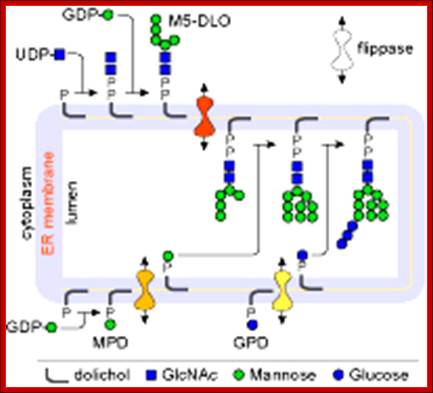
Lipid flip-flop across the ER during assembly of the oligosaccharide-PP-dolichol precursor of protein N-glycans. Glc3Man9GlcNAc2-PP-dolichol, the glycan donor in protein N-glycosylation, is synthesized in a multi-step pathway in the ER. The first 7 steps, leading to the synthesis of M5-DLO from dolichol-P occur on the cytoplasmic face of the ER. GDP-mannose and UDP-GlcNAc directly contribute the sugar moieties for these reactions. M5-DLO is then flipped into the ER lumen by M5-DLO flippase, and extended in a further 7 reactions to yield Glc3Man9GlcNAc2-PP-dolichol. The sugar donors for these reactions are (mannose-phosphate dolichol) MPD and glucose-phosphate dolichol (GPD). MPD is synthesized from dolichol-P and GDP-mannose on the cytoplasmic face of the ER, then flipped to the ER lumen by MPD flippase. In the lumen it is the mannosyl donor for the 4 reactions that convert M5-DLO to Man9GlcNAc2-PP-dolichol. GPD is synthesized from dolichol-P and UDP-glucose on the cytoplasmic face of the ER, then flipped to the ER lumen by GPD flippase. http://www.cornellbiochem.org

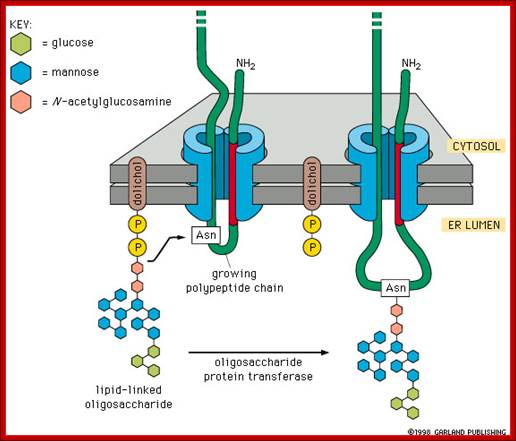
Glycan assembly and attachment: Precursor glycan synthesis begins on the cytosolic face of the endoplasmic reticulum (ER) and is completed after the structure is flipped into the ER lumen. Oligosaccharide transferase (OSTase) then transfers the precursor glycan to the Asn residue on the nascent protein. http://www.piercenet.com/browse.cfm?
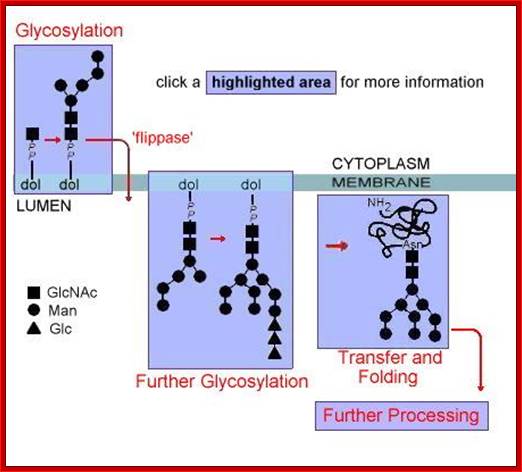
http://slideplayer.com
One aspect to note is that not all Asn residues with the predicted consensus sequence are glycosylated. N- to C-terminal protein synthesis results in transport of the growing polypeptide into the ER in the same orientation, and protein folding occurs soon after the polypeptide enters the ER. Therefore, as protein folding increases, OSTase is less able to access the consensus sequence for glycan transfer. Indeed, more N-terminal Asn residues are glycosylated than C-terminal Asn residues.
Note stepwisae transfer of Dolichol phosphate with glycans are flipped to ER lumen and then then the glycan group is transferred on to asn position of the ER protein. The Dolichol multimer is flipped back to Cytosolic surface.
http://biochem4.okstate.edu/~firefly/Bioch205/Bioch205clmfolder/b205clm23/proteinsynthesis.htm
The pathway of N-linked protein glycosylation is conserved among most of the eukaryotic organisms. This conservation made it possible to use the model organism S. cerevisiae to characterize the complex pathway by applying yeast genetics and biochemical techniques. N-linked protein glycosylation initiates with the assembly of an oligosaccharide on a lipid-carrier, first on the cytoplasmic side of the endoplasmic reticulum (ER) membrane, catalyzed by a series of specific glycosyl transferases. The resulting intermediate is flipped into the ER lumen where more hexose residues are added by a distinct set of glycosyl transferases. The oligosaccharide is finally transferred en bloc from the Dolichol lipid to selected asparagine residues of secretory proteins by the key enzyme of the pathway, the oligosaccharyl transferase (OST).
· Endoplasmic reticulum and Golgi complexes contain transporters, for nucleotide sugars, as carriers. Dolichol-p-mannose and Dolichol-p-glucose provide the needed mannose, glucoses and others as independent components. Once their mannose or glucoses are transferred from Dolichos~P~P, the Dolichol-Ps’ are flipped back towards cytosolic surface, where they are again regenerated as Dolichol-p-p-mannose and Dolichol-p-p-glucose etc.
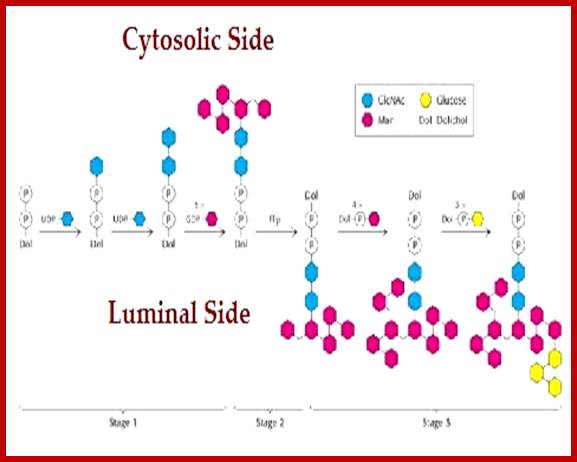
Addition of Glyco components to Dolichol-P~P (pyrophosphate) starts at cytosolic side of ER. Once the basic carbohydrate complex or tree is built it is flipped towards the luminal side of the ER; where more carbohydrates are added and modified. Such carbohydrate complexes are specific-to-specific proteins and they are linked at specific sites of the protein in sequence specific and motif dependent manner. All the required enzymes and components are found in cytoplasm and in ER lumen. Once the components are built at cytosol they are then flipped towards lumen side. Some of the enzymes called flippases are responsible flipping the dolichol linked glycol groups from cytosolic side to ER luminal side. https://www.slideshare.net/
Once the dolichol glycosyl clusters are inside the ER lumen, the more mannose are added and three more glucoses are added; this cluster is transferred to Asn site of a protein found in the ER or a protein that is just transferred into the lumen; http://quizlet.com/
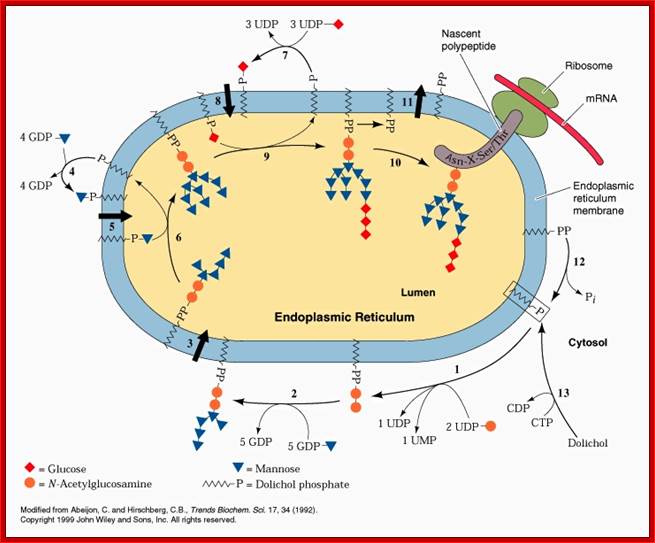
www.slideplayer.es/slide
Dolichos-~P-P gets laid with 7 glycans, then it is flipped by Flippase found in the membrane; there it further goes through addition of different glycans in a set pattern. Once the glycosylation is complete, the glycol-tree is transferred to the incomming nascent protein at Asn. The Dolichol phosphate is flipped back to assemble another glyco branch.
Asn (N) linked Glycosylation; https://calldutyblack.ru/
A list of Glyco Transporters:
Glyco’s units are transported across the ER membrane by specific nucleotide transporters, -a list is given below.
UDP-N- acetyl glucosamine,
UDP-N acetyl Galactosamine,
UDP-N-acetyl muramic acid,
UDP-n acetyl Neuraminic acid,
UDP-Galactose,
UDP-glucose,
UDP-Glucuronic acid,
UDP-Xylose,
GDP-Fucose,
GDP-mannose,
CMP-Sialic acid,
The O-linked glycosylation:
· Mucins, a sub-maxillary gland product, have O-linked glycosylation. In this, serine or threonine (hydroxyl amino acids), are added with glycols. Most of them are secreted.
· The site specificity is determined by its structural motif of the protein containing serine or threonine.
· All blood group proteins, which elicit antibody responses, if they are not self, are serine-O-linked glycoproteins. Many of the ser-linked glyco-proteins are secreted through saliva, they are also found in Zona pellucida, and they act as receptors for recognizing specific sperm types.
· Addition of carbohydrates is a stepwise process; most of it takes place in Golgi luminar space for they contain required enzymes.
· As in other cases the carbohydrate carriers are UDPs.
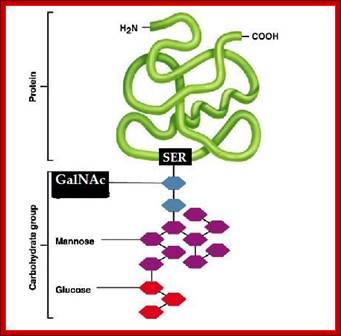
The simple diagram shows O-linked Glycosylation. www.suggest-keywords.com
While N-glycosylation is the most common glycosidic linkage, O-glycoproteins also play a key role in cell biology. This type of glycosylation is essential in the biosynthesis of mucins, a family of heavily O-glycosylated, high-molecular weight proteins that form mucus secretions. O-glycosylation is also critical for the formation of proteoglycan core proteins that are used to make extracellular matrix components. Additionally, antibodies are often heavily O-glycosylated.
O-glycosylation occurs post-translationally on serine and threonine side chains in the Golgi apparatus. N-glycosylation does not preclude the other from occurring, as O-glycosylation commonly occurs on glycoproteins that were N-glycosylated in the ER. Besides the different linkage, O-glycosylation also differs in the method of glycosylation.
While a precursor glycan is transferred en bloc to Asn via N-glycosylation, sugars are added one-at-a-time to serine or threonine residues. O-glycosylation can also occur on hydroxylysine and Hydroxy proline, oxidized forms of lysine and proline respectively that are found in collagen. Additionally, O-linked glycans usually have much simpler oligosaccharide structures than N-linked glycans, Protein Methods Library
· In the case human ABO blood group antigens the first carbohydrate that is added to serine is N-Acetyl Galactosamine (N-Ac-GAL-amine). Then they are added with N-Ac-Glc (Glc=Glucose), GAL (Galactose) and Fucose in that order. This is precursor glycol cluster. But this glycol-cluster is modified in different human genotypes as O-group, A-group and B-group by specific transferases called A and B types. The antigen precursor is modified by O or H enzyme to produce O-group antigens and the O-group antigens are can be further modified into A-group and B-group by A-enzymes and B-enzymes respectively.
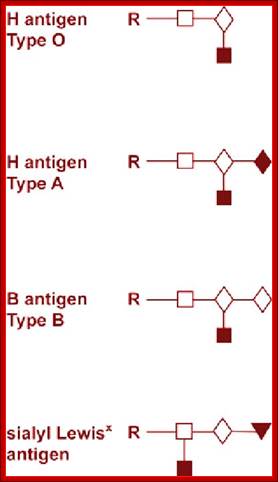
A, B and O Antigens’ Glycosylation
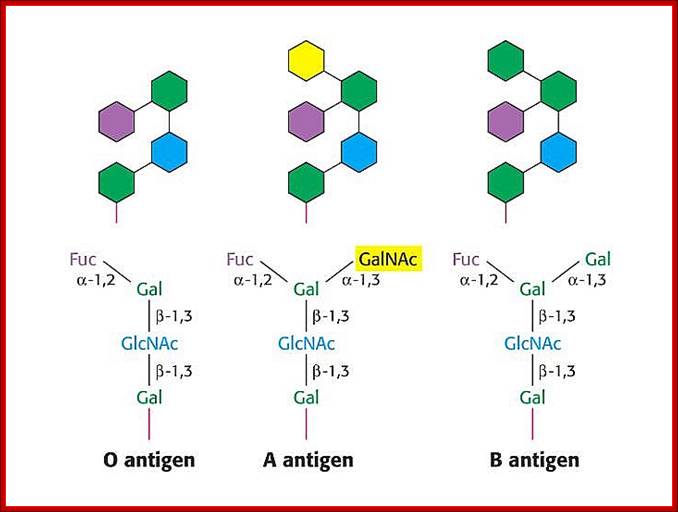
Blood group antigenic Glycoproteins; https://www.slideshare.net
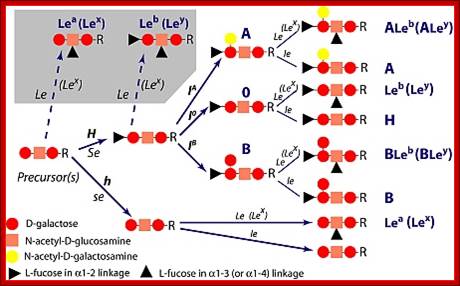
Lewis antigens- Sub groups; http://www.ncbi.nlm.nih.gov/
· The O-group contains- protein-ser-O-NAc-GAL-amine-GAL-NAc-Glc-GAL-Fucose linked in the same order. This is performed by O- or H-enzymes. In A-genotype, the linkage is- protein-ser—O-NAc GAL amine-GAL-NAc Glc-GAL-NAc-GAL amine, Fucose is found at GAL as it is found in O antigen. The human blood group B-type has – protein-ser-O-NAc
· GALamine-GAL-NacGlc-GAL-NAcGALamine, the Fucose is linked to GAL as in the O-group antigen.
Most of the cytosolic proteins are devoid of glycosylation, but some proteins in nuclear pore complex and some transcription factors contain N-linked glycosylation (NAG) at a specific site. At another site hydroxylysine is glycosylated with single Galactose or with two Glucose-Galactose residues. Arabinose is a rare addition to hydroxyl-proline in yet another site.
Glycosyl-Phosphotidyl Inositol (GPI) linked Proteins (Glypication):
· Glycosyl Phosphotidyl Inositol (GPI) acts as an anchor for a wide variety of extra cellular proteins localized at extra cellular surface of the membranes. The core of GPI structure is synthesized in the luminal side of ER and it is found mostly in Golgi lumen fluid but anchored.
· First, Di-acyl Phospho glycerol is added to 1, 4, 5 triphosphate-Inositol, and then an N-Acetyl Glucosamine is added to tri phospho-Inositol. The three to four mannoses are added to NAG (N-Acetyl Glucosamine), and the carrier molecule for mannose is Dolichol-phosphate.
Every time it carries the mannose from cytosolic side and flips towards the luminal side and donates a single mannose and flips back. The last to be added is ethanolamine.
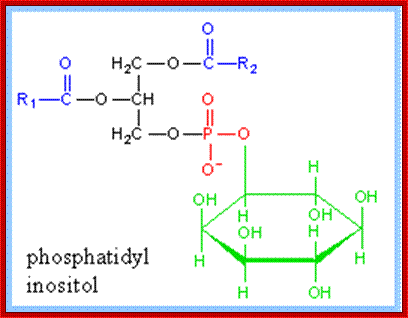
Phosphatidyl (PI) Inositol: https://www.rpi.edu
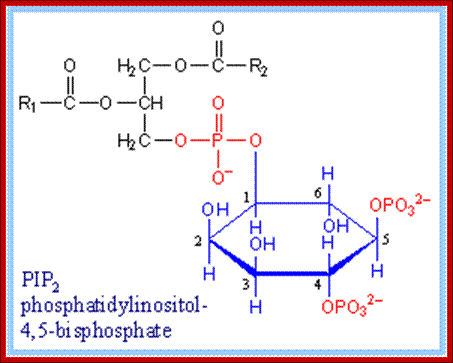
Phosphotidyl inositol1, 4, 5-Phosphate; https://www.rpi.edu
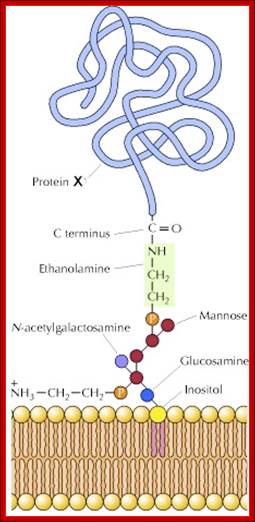
Glyco-phosphatidylinositol anchored to the membrane via lipid components. http://celladhesionlab.com/
Phosphoethanolamine group links to COO (-) group of the terminal part of the protein (with some amino acids removed) www.cineskane.se
[NH2.CH2.CH2-PO4-Man-Man-Man-Man-GalNH2-O-IP1-PO4-OCH2-CHoAcyl)-Ch2-O-acyl group]
· Membranes do contain specific carbohydrate transporters into the core of ER lumen. They exist as complexes bound to membrane anchors.
· GPI s are formed inside the lumen and they are also present in the cis or mid golgi membranes
· When a particular protein is identified, the NH2 group of the ethanolamine nucleophilically attacks a specific (structure dependent) aminoacyl group at the C- terminal part of the protein, to form a –C (=O)-NH- bond between Glycine and ethanolamine of GPI. This releases 20 to 30 amino acids from the C- terminal part of the protein. This GPI-anchored protein is transported across the lumen; and it is ultimately transported to the exterior surface of the cell and still anchored to the exterior surface of cell membranes.
Most of the GPI anchored proteins are found as cellular receptors, some found in immune proteins, many are found anchoring enzyme to membranes. Some of the GPI linked proteins are secreted.
Glycation:
Glycation is non-enzymatic attachment of reducing sugars to the nitrogen atoms of proteins (both to the N-terminus and to lysine and histidine side chains). This type of modification is also known as the “Maillard” reaction. Over time, the sugar moieties bound to Glycated proteins are gradually modified to become Advanced Glycation End products (AGEs), some of which are implicated in a variety of diseases, such as type II diabetes mellitus, cancer, atherosclerosis, Alzheimer disease and Parkinson disease.
C-Mannosylation:
A mannose sugar is added to the first tryptophan residue in the sequence W-X-X-W (W indicates tryptophan; X is any amino acid). Thrombospondins are one of the most commonly C-modified proteins, although this form of glycosylation appears elsewhere as well. C-mannosylation is unusual because the sugar is linked to a carbon rather than a reactive atom such as nitrogen or oxygen. Recently, the first crystal structure of a protein containing this type of glycosylation has been determined - that of human complement component 8, PDB ID 3OJY (WIKI).
Phosphoglycation:
This type of post-translational modification is limited to parasites (e.g., Leishmania and Trypanosoma) and slime molds (e.g., Dictyostelium) and is characterized by the linking of glycans to serine or threonine via phosphodiester bonds. In some parasitic species, such as Leishmania, phospho glycosylation is the most abundant post-translational modification and is used to make proteophosphoglycans (PPGs), which are critical for protection against host complement and promote parasite aggregation in the host. Similar to N-glycosylation, phospho glycosylation occurs by transfer of a prefabricated phosphoglycan from a membrane-bound molecule via a phosphor-glycosyl transferase (PTase), although the exact structure and enzyme varies by species.
Post-Glycation Modifications:
Besides multiple types of glycosylation occurring on the same protein, glycans can be further modified to increase the diversity of glycoproteins in a given proteome. These modifications include:
- Sulfation at Man and GlcNAc residues in the production of glycosaminoglycans (GAGs), which are components of proteoglycans in the extracellular matrix
- Acetylation of sialic acid to facilitate protein-protein interactions.
- Phosphorylation, such as with Man residues on precursor lysosomal proteins (mannose 6-phosphate) to ensure trafficking to lysosomes by binding to mannose 6-phosphate receptor (M6PR) in the Golgi.
Proteins Anchored to Lipid Chains:
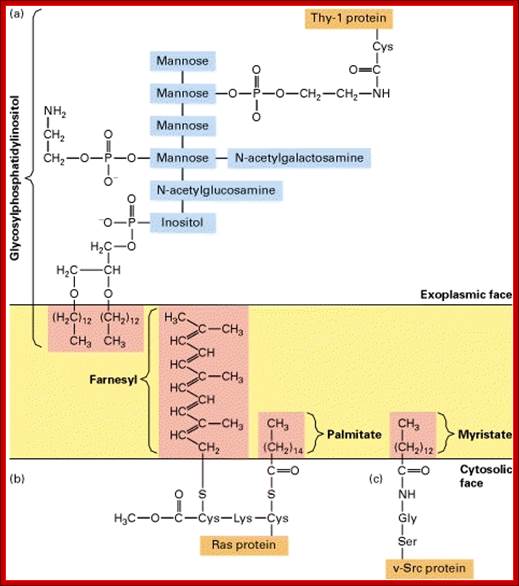
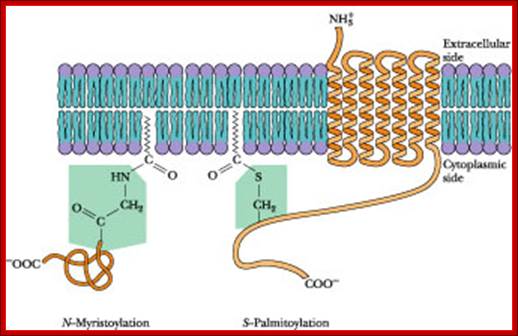
· In many cases the proteins, either at N-terminal region or at C-terminal region, are linked to lipids anchored in membrane components; such as Farnesyl molecules (15C), Geranyl (10C) components (isoprene derivatives) or Steroyl (18C) or Myristoyls (14C) or Palmitoyl (16C) and in many cases proteins are attached to membrane anchored GPI (Glycosyl Phosphotidyl Inositol complex).
· Myristoyl (14C) COO^- group: It can be linked to an amino terminal of a protein; a myristoylated protein is mostly localized at cytosolic surface.
· Palmitoyl (16C) COO^- group: It can be linked to the OH group of any one of the internal serine or threonine of proteins, again localized at cytosolic surface, ex. cAMP dependent kinase.
· Steroyl (18C) COO^-: It can be bonded to SH group of any one of the internal cysteine and the protein is localized at cytosolic surface.
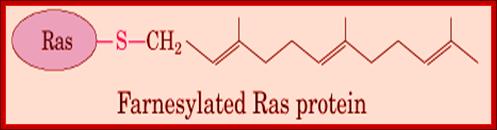
· Farnesyl (15C): It can be linked to cysteine at carboxy terminal of a protein (localized at cytosolic surface).

Palmitoyl, Myristoyl ation to membrane proteins; http://what-when-how.com
http://what-when-how.com/molecular-biology
Some of the fatty acid chains used as anchors in the membranes for specific proteins.
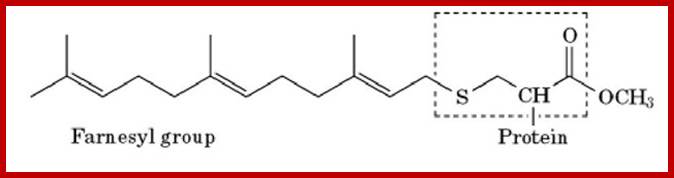
Farnesylation is the process by which a cysteine residue in the C-terminal region of some eukaryotic proteins is posttranslationally modified with an isoprenoid lipid (the 15-carbon farnesyl group) and the exposed carboxyl group is methylated (Fig. 1). The farnesylation and carboxylmethylation increase the affinity of the protein for the membrane and have important functional consequences. The farnesyl group is one of several lipids that act as a membrane anchor for proteins. The role of the farnesyl group and other lipids in membrane anchoring is described in more detail elsewhere [see Prenylation; Membrane Anchors]
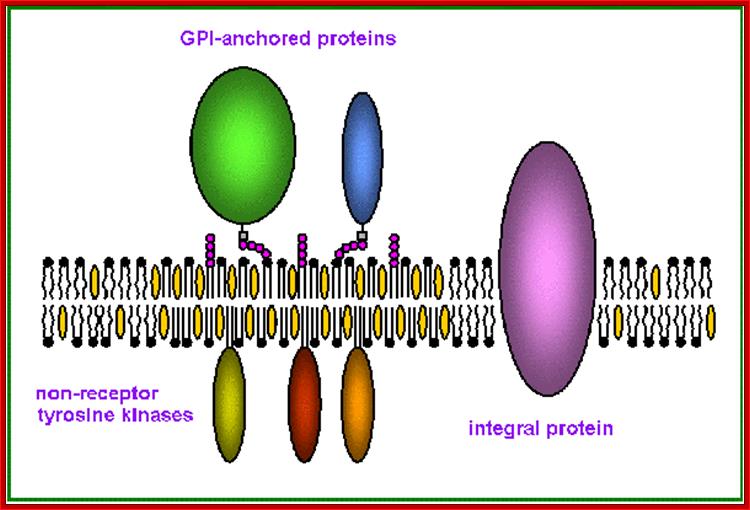
Proteins anchored to GPI are found at the exoplasmic surface and the proteins linked to certain fatty acids are found at cytosolic surface
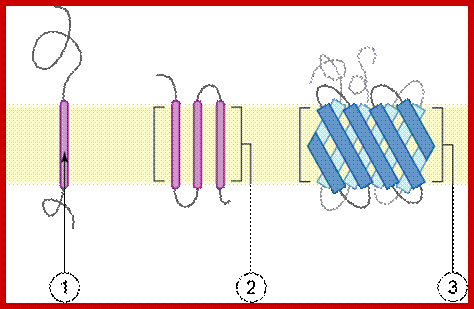
Intrinsic proteins- A transmembrane protein is a protein that spans the entire biological membrane. Transmembrane proteins aggregate and precipitate in water. They require detergents or nonpolar solvents for extraction, although some of them (beta-barrels) can be also extracted using denaturing agents. http://flipper.diff.org
![Image result for Prenylation; Membrane Anchors]](Co_and_Post_Translational_Events4-Glycosylation_of_Proteins_files/image068.jpg)
http://www.allometric.com
Anchoring of integral proteins to the plasma membrane by membrane-embedded hydrocarbon groups (highlighted in red) (a) protein and several hydrolytic enzymes are anchored by glycosylphosphatidylinositol. This complex anchor is found only on the exoplasmic face. (b) Cytosolic proteins involved in signaling such as Ras are anchored to the cytosolic face of the membrane through farnesyl and palmitoyl groups. (c) Other cytosolic proteins are associated with the membrane through myristate and similar fatty acids attached to an N-terminal glycine residue.
Topology of membrane proteins
http://flipper.diff.org
Fatty acid-S anchored membrane proteins
http://www.sciencedirect.com
Proposed structure and organization of a lipid raft microdomain in the plasma membrane. Sphingolipids, which include both sphingomyelin and glycosphingolipids, associate with cholesterol to form a more tightly packed domain. The regions rich in phosphatidyl choline and other glycerol-based phospholipids are less densely packed, and form fluid regions outside the raft micro domains. Lipid rafts are enriched in GPI-anchored proteins and enzymes at their external surface, and acylated proteins, such as tyrosine kinases of the Src family (p56lck, p60fyn, and p53/56lyn etc) at the cytoplasmic surface. Transmembrane integral proteins are generally excluded from rafts, and are found in the more fluid phospholipid-rich regions of the membrane

Glycan region of GPI anchored-protein; Yuich Abea et al; http://www.sciencedirect.com
The above said lipid-fatty acid chains act as anchors for proteins, some of which may be disposed to the outer surface and some towards cytosolic surface. Specific transferase enzymes perform these linkages. Most of the cellular lipids are synthesized at the cytosolic surface of ER and in the lumen of ER and the enzymes responsible for synthesis or localized or anchored to membranes and their enzymes are found at both cytosolic and Luminal surface.
Quite a number of proteins as they are modified or processed are hooked on to many lipid derivatives that are anchored to membranes. Some proteins are prenylated to carboxyl group of the protein. These proteins use cysteine at carboxyl end and link to carboxyl group of the Farnesyl group (15C chain) or Geranyl-geranyl group (20C chain) of molecules. A very good example of such membrane linked protein is monomeric Ras proteins and gamma subunit of Transducin in the eye. Even sarcoma protein is anchored to the membrane through such prenylation. They use carboxyl end sequence such as CAAX (Cysteine, Alanine, Alanine and X is any amino acid), where SH- group of the cysteine is covalently linked to the Carboxyl end of the prenyl chain.
· Addition of glycosyl residues induces proper folding. It also requires PDIs and help of chaperones (BiPs).
· Endoplasmic resident proteins contain KDEL as marker for the retention of the luminal proteins within ER. Whichever proteins contain these sequences they are retained in ER.
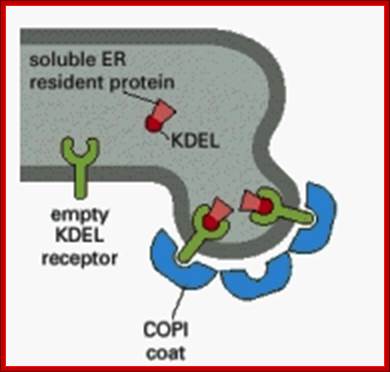
KDEL receptors anchored to ER membranes bind to proteins that have KDEL signatures. http://quizlet.com/
- The ER is also involved in cell signaling by releasing stored calcium ions (Ca2+) into the cytosol. This serves to amplify signals from molecules in very low concentrations, such as extracellular hormones, thus triggering a response in cells. Without amplification of the initial signal, the cells will not respond to the hormone. The chain of events can be summarized as follows: (1) a hormone binds to a specific receptor on the plasma membrane; (2) the receptor interacts with several other membrane-bound signaling proteins to produce a molecule called inositol-1, 4, 5-trisphosphate (IP3), which is released into the cytoplasm; and (3) IP 3 interacts with its own receptor on the ER membrane and passes the signal on to a Ca 2 pump. Once the pump is activated, it releases a massive amount of calcium from the ER lumen into the cytoplasm. The Ca 2 ions act as "second messengers" that turn on several cellular systems ranging from cell motility to protein synthesis.

Glycan maturation. After initial trimming in the ER, the glycoprotein is trafficked to the
Golgi, where Golgi mannosidase I removes multiple mannose sugars. Glycans that do not undergo further glycosylation are called high-mannose oligosaccharides. Further sugar addition and removal yields a common core oligosaccharide onto which multiple Gtfs add different sugars to generate the highly variable complex oligosaccharides. Glycan maturation beyond the common core provides endo H insensitivity. Glycans can be high-mannose, complex or a combination of both (i.e., hybrid oligosaccharide). Protein Methods Library; http://www.piercenet.com/method/protein-glycosylation.
· After the proteins are added with oligo clusters, processing starts within the Rough Endoplasmic Reticulum-RER itself, this is even before they are transported into Smooth Endoplasmic Reticulum-SER. The SER region is involved in the synthesis of Fatty acids and phospholipids. From SER glycol-proteins are transferred to the Cis surface of the Golgi that is proximal to SER and the other end of the Golgi complex is called Trans Golgi. The vesicles are formed and pinched of by a group of proteins called COPII (Coatamer Proteins II). It is at cis surface, the Golgi membranes form by fusion of ER-SER vesicles loaded with glycosylated proteins. Such vesicles fuse with one another aided by specific membrane proteins and form flat sheath like structures; thus the vesicle fused sheets stack one above the other. The other or distal end, called Trans Golgi, membranes leave from the Golgi as vesicles loaded with processed and assorted proteins destined to different destinations.
· In the Cis and Median and Trans regions of the Golgi complex proteins are subjected further modifications such as removal of certain monosaccharaides and addition of few more monosaccharaides at different positions to final product. In most of the cases each and every kind of proteins that are modified are marked, similar to lysosomal proteins. This greatly helps in sorting of different proteins.
· The matured proteins in trans-Golgi ready to be loaded into trans-golgi vesicles. By the time proteins reach this stage they are matured, sorted and loaded into trans-glogi vesicles and budded off.
· For example one of the non-lysosomal protein that is transferred to Cis region contains N-linked oligosaccharide clusters with 2 NAG and 9 mannoses, with five mannoses in one of the branched branches and three more mannose in another branch. In the Cis region one mannose is removed from one of the mannose branch to produce a six-mannose group. This forms what is called inner core group. Then from the protein in medial region, some mannoses are removed and two additional branches are produced with N-acetyl Galactose added to the terminal mannose residues and one Fucose is also added to one of the two NAGs. Then the protein in medial Golgi matures into Tran’s region where more modifications result in the formation of two mannoses as branches. From one of the mannose branch two NAGs are added as branches. The other mannose is also added with NAG. Then each of the free NAGs is added successively with one Galctose and one NANA (N acetyl neuraminic acid). An antibiotic Tunicamycin, whose structure is similar to that of UDP-glucosamine, inhibits Glycosylation of proteins. Backtracking is another inhibitor of glycosylation, for it binds Dolichol-PP and inhibits dephosphorylation.
· Most of the lysosomal enzymes have N-linked glycosylations. In order to identify from other cellular proteins, one of the mannose residue is phosphorylated at its 6-C position as mannose-6-phosphate. This acts as lysosomal marker and this marker is used by specific receptor anchored in luminal side of ER and facilitates the transport to Golgi complex from cis to mid Golgi and then to trans Golgi. Similarly different proteins destined to specific targets may have such markings, so as to facilitate assortment and transport. On the luminal side of the ER or Golgi membranes there are receptors for mannose 6-P anchored to membranes for binding to lysosomal proteins.
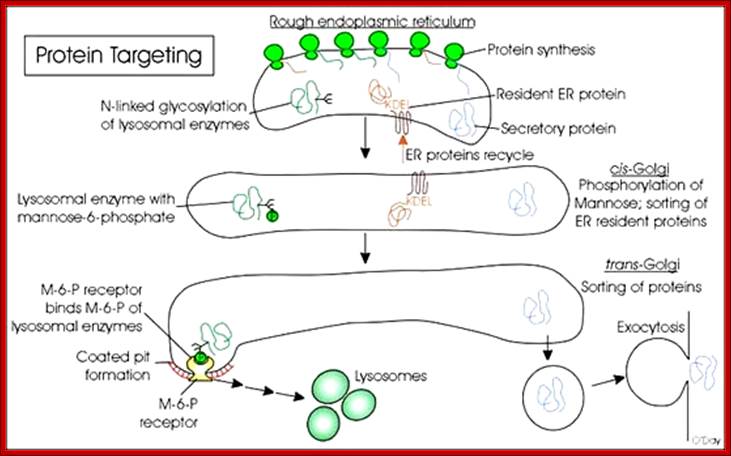
Sorting of proteins using specific signal sequences and receptors; www.utm.utoronto.ca
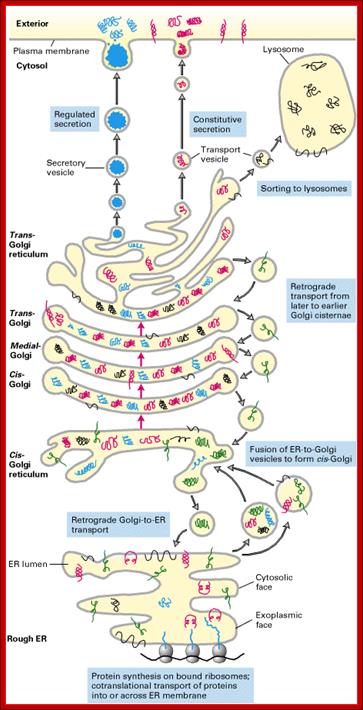
http://www.unitus.it/
The secretory pathway of protein synthesis and sorting. Ribosomes synthesizing proteins bearing an ER signal sequence become bound to the rough ER. As translation is completed on the ER, the polypeptide chains are inserted into the ER membrane or cross it into the lumen. Some proteins (e.g., rough ER enzymes or structural proteins) remain resident in the ER. The remainder move into transport vesicles that fuse together to form new cis-Golgi vesicles. Each cis-Golgi cisterna, with its protein content, physically moves from the cis to the trans face of the Golgi stack (red arrows). As this cisternal progression occurs, many luminal and membrane proteins undergo modifications, primarily to attached oligosaccharide chains. Some proteins remain in the trans-Golgi cisternae, while others move via small vesicles to the cell surface or to lysosomes. In certain cell types (e.g., nerve cells and pancreatic acinar cells), some soluble proteins are stored in secretory vesicles and are released only after the cell receives an appropriate neural or hormonal signal (regulated secretion). In all cells, certain proteins move to the cell surface in transport vesicles and are secreted continuously (constitutive secretion). Like soluble proteins, integral membrane proteins move via transport vesicles from the rough ER to the cis-Golgi and then on to their final destinations. The orientation of a membrane protein, established when it is inserted into the ER membrane, is retained during all the sorting steps: Some segments always face the cytosol; others always face the exoplasmic space (i.e., the lumen of the ER, Golgi cisternae, and vesicles or the cell exterior). Retrograde movement via small transport vesicles retrieves ER proteins that migrate to the cis-Golgi and returns them to the ER. Similarly, cis- or medial-Golgi proteins that migrate to a later compartment are retrieved by small retrograde transport vesicles. [See B. Glick and V. Malhotra, 1988, Cell 95:883.]; http://www.unitus.it/
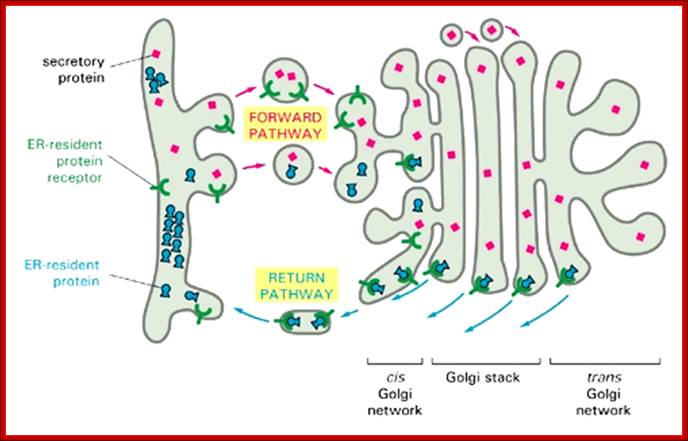
Figure: The mechanism used to retain resident proteins in the ER; https://www.slideshare.net
ER-resident proteins that are transferred to the cis Golgi network are returned to the ER by vesicular transport. A membrane receptor in the cis Golgi network captures the proteins and carries them in transport vesicles back to the ER. The ionic conditions in the ER dissociate the ER proteins from the receptor, and the receptor is then returned to the cis Golgi network for reuse. Receptors for the ER retention signal are also found in the cis, medial, and trans Golgi cisternae. Thus the retrieval of ER proteins begins in the cis Golgi network, but the return pathway operates from the later Golgi cisternae as well. The retention is aided by interactions between ER-resident proteins in the ER lumen. These interactions retard the exit of ER proteins relative to proteins that are destined for secretion (secretory proteins). Experiments in which the ER retention signal is removed from BiP show that BiP leaves the ER and is eventually secreted from cells but that its exit from the ER occurs much more slowly than that of bona fide secretory proteins, indicating that it is held there by weak interactions with other proteins.
General Features and Functions of Glycoproteins:
Proteins are also modified by acylation, phosphorylation, methylation, sulfation, and hydroxylation in sequence specific manner. There are innumerable proteins, which are modified by various methods and means for different functions. Here some examples have been given with general features.
1. The CD4 receptor proteins on T-lymphocytes contain one or two N-linked glycosylations; if glycosylations fail the proteins don’t fold properly, yet they are transferred to Golgi.
2. Sucrase-isomaltase enzymes are found in microvilli as glycocalyx, they contain many sugar residues. Glycocalyx is a loose network of glycoproteins and glycolipids.
3. Glycoproteins act as chaperones, ex. Calnexin and Calreticulin.
4. They absorb water and give good solubility environment.
They act as carriers-ex. Ceruloplasmins carry iron to serum.
5. IgG- in human carry glycosylation clusters in one arm of the antibody and also in the stem region of heavy chain. All in all they contain 30 N-linked oligosaccharides.
6. RNase B contains N-linked 5-6 mannose residues in two or more sites.
7. GM-CSF: Granulocyte and Macrophage cell stimulating factors have 2 N-linked glycosylations and 5 O-linked glycosylations. They act as carriers-ex. Ceruloplasmins carry iron to serum.
8. IgG- in human carry glycosylation clusters in one arm of the antibody and also in the stem region of heavy chain. All in all they contain 30 N-linked oligosaccharides. Vasoactive intestinal peptides (VIP receptors) contain 2 –3 N-linked sialated oligosaccharides .
9. Human Aids virus gets entry into cells by receptor proteins, which are glycoproteins called chemokine receptors.
10. Poxy virus binds to chemokines and enters cells.
11. Gastric mucous cell surface glycoproteins provide surface for infection of Helicobacter pylori which causes gastritis and gastric and duodenal ulcer.
12. Similarly adenoma cancer causing bacteria binds to glycoproteins of gastric mucous cells.
13. Rabies viruses bind to neuronal cell adhesion molecules such as N-CAM.
14. Herpes virus infects through fibroblast receptors for growth factors.
15. GPI anchored proteins are found on the surface of erythrocytes. Such proteins are also linked to acetyl esterase, alkaline phosphotase, and 5’Nucleotidase, N-Cam, T-cell markers such as Thy-1, lymphocyte antigen called LFA-3 (lymphocyte function associate antigen).
16. Hydroxylation of proteins at lysines and prolines sites is vitamin-C mediated; examples are collagens, oxytocin and vasopressin.
17. In some glutamic acid of a protein is modified by carboxylation to generate γ-carboxy glutamate, which is mediated by Vitamin-K. This protein is found in blood clotting cascade.
Impact of Glycoproteins:
Glycoproteins, Glycolipids and Proteoglycans play essential roles in various biological phenomena such as development, immune response, and tissue functions. Several human congenital diseases have proven to be defects in the glycosylation of proteins. Infectious diseases and immune response are dependent on carbohydrate residues. There are profound neurological consequences to humans suffering from diminished glycosylation found in chronic alcoholism and ageing. Structural studies at this level provide the potential to reveal new targets for drug intervention
Many biological processes manifest themselves at the level of protein activity. The rapid accumulation of genomic sequence data has driven the development of new methods for recognizing protein function, protein interactions and changes in expression patterns. Until recently, however, rapid and reliable protein identification with high sensitivity was not possible. Recent developments in mass spectrometry have created an ongoing revolution in proteomics, enabling the identification of proteins with the sensitivity needed to address important biological problems.
Proteoglycans:
Proteoglycans are proteins that are heavily glycosylated. All proteoglycans have a core protein with greater amount of characteristic glycosaminoglycan (GAG) chain(s) attached. The carbohydrate chains are long, linear polymers that are negatively charged under physiological conditions, due to the occurrence of sulfate and uronic acid groups. Proteoglycans occur in the connective tissue.
Based on the kind of Glycosamine-Glycans (GAGs) and the numbers added to protein core they are classified into different kinds.
A list of Proteoglycans (PG) (small and large complexes):
The following are different types of PGs- Aggrecan, Versican, Syndecan, Serglyci, Perlican, Bamacan, Glypican, Testican, CD44, XVIII Collagen, Decorin, Collagen IX, Anechorin, Biglycan, Lumican, Fibromodulin, Neurocan and Brevican (list incomplete).
Glycosamino-Glycans (GAGs) basic glycan polymers:
There are a group of long polysaccharide chains built out of disaccharide units. They can exist as extra cellular material or they can be linked to protein threads, which in turn are linked to a poly-hyaluron polymer of high molecular weight.
The di-glycan subunits can polymerize into long chains of high molecular weight aggregates called Glycosamino-Glycans (GAGs). The combination of monosaccharides varies from one GAG to another GAG. One of the two repeating units is always N-acetyl glucosamine or N-acetyl galactosamine. The other one is mostly D-Uronic acid or L-Uronic acid. They are variously modified; mostly amino sugars are esterified with sulfates. They are secreted out of cells and always found in extra cellular space of connective tissues. They hold cells together, provide space for diffusion, they are found as interlocking meshwork of hetero polysaccharides, fibrous proteins such as fibronectins, collagens, elastins, lamins and others. They absorb plenty of water, swell and can withstand shocks and intense pressures, like shock absorbers.
Hyaluronate: Glucuronate-N-Acetyl Glucosamine linked to by β1-3 linkage. There can as many as 50,000 disaccharide pairs. It is transparent, viscous fluid like, acts as lubricant in synovial fluid in joints, vitreous humor, extra cellular matrix of cartilage and tendons. It absorbs plenty of water; and absorbs pressure and shocks. It is this that facilitates smooth movement of knees, and other joints. If this fluid is damaged one suffers from joint pains; a life long suffering.

Hyaluronate; http://www.naturalingredients.ru/
Chondroitin sulfate: D-Glucuronate linked to N-acetyl galactosamine by β1-3 linkage; at 4’ or 6’-galactosamine is sulfated. They produce 20-60 pairs of long chains. This is found abundantly in cartilage, tendons, and ligaments and in the walls of aorta.

Comparison of two structures with strong binding capability to midkine ; http://www.glycoforum.gr.jp
Keratan sulfate: D-Gal β1-4 linked to N-Acetyl-Glucosamine with 6-CH2SO4. It can form 25-30 disaccharide pairs. Found in cornea, cartilage, bones, dead horns, hairs, hoofs, nails and claws.

Proteoglycans ;Keratan Sulphate; https://www.slideshare.net/
Dermatan Sulfate: D-Iduronate linked to N-Acetyl Galactosamine-sulfate (beta 1-> 3 linkages). Sulphate is then added at 4C’. Most of them are found as 20-24 pairs of GAGs. These are found in skin, heart valves and blood vessels.
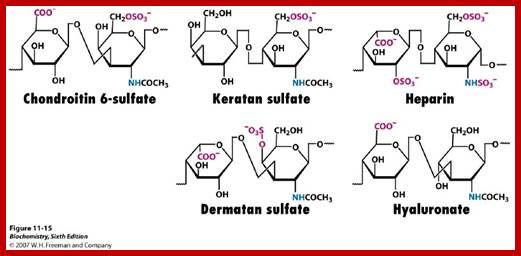
http://oregonstate.edu
Heparan SO4: D-Glucuronate is β1-4 linked N-Acetyl Glucosamine-and sulfate at 6th position. Found in basement membrane, cell surface, contain high amount of acetylation, its other form Heparin is mostly found in mast cells, lining arteries of lungs, liver and skin. Heparin with out sulfate can also exist.
It is important to note that some polysaccharides especially oligosaccharides mentioned above are a kind of information carrier molecules. Act as destination labels for some proteins. Act as mediators for cell-to-cell recognition and interaction. Act as molecules to go in between cell and extra cellular matrix. Some of them involve in cell to cell adhesion and cell migration, blood clotting, and immune response and wound healing. All of them are a kind of information molecule, and they also found linked to protein cores or lipids as proteoglycans and glycolipids.
Proteoglycans and GAGs are generally secreted out of the cells, and they are found enmeshed with other extra cellular glycoproteins such as collagens, fibronectins, elastins and other glycoproteins. They generate large volumes and with negative surfaces charges. They can hold large amount of water because of hydrating properties. The can be compressed without much of damaged to them and withstand high compressions. They can function as lubricants and synovial liquid for easy movement of joints; they provide filter like space for movement of cellular components and even cells squeeze through them. They also provide extra strength and resilience at extra cellular space, it also provide extra strength to other extra cellular molecules. They are also involved in presenting hormones to specific receptor or they may be involved in ligand binding. Age can detiorate these liquids.
The most abundant hetero-polysaccharides in the body are the glycosaminoglycans (GAGs). These molecules are long unbranched polysaccharides containing a repeat of disaccharide units. The disaccharide units contain either of two modified sugars, N-acetylgalactosamine (GalNAc) or N-acetyl glucosamine (GlcNAc), and a uronic acid such as glucuronate or iduronate. GAGs are highly negatively charged molecules, with extended conformation that imparts high viscosity to the solution. GAGs are located primarily on the surface of cells or in the extracellular matrix (ECM). Along with the high viscosity of GAGs comes low compressibility, which makes these molecules ideal for a lubricating fluid in the joints. At the same time, their rigidity provides structural integrity to cells and provides passageways between cells, allowing for cell migration. The specific GAGs of physiological significance are hyaluronic acid, dermatan sulfate, chondroitin sulfate, heparin, heparan sulfate, and keratan sulfate. Although each of these GAGs has a predominant disaccharide component (see Table below), heterogeneity does exist in the sugars present in the make-up of any given class of GAG.
Hyaluronan is unique among the GAGs in that it does not contain any sulfate and is not found covalently attached to proteins as a proteoglycan. It is, however, a component of non-covalently formed complexes with proteoglycans in the ECM. Hyaluronic acid polymers are very large (with molecular weights of 100,000–10,000,000) and can displace a large volume of water. This property makes them excellent lubricators and shock absorbers.”
|
|
Hyaluronates:composed of D-glucuronate +
GlcNAc |
|
|
Derma tan sulfates:composed of L-iduronate (many
are sulfated) |
|
|
Chondroitin 4- and 6-sulfates :composed of D-glucuronate |
|
|
Heparin and Heparan sulfates:composed
of iduronate-2-sulfate (D-glucuronate-2-sulfate) |
|
|
Keratan sulfates:composed
of galactose + GlcNAc-6-sulfate |


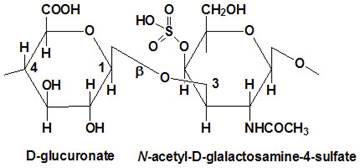
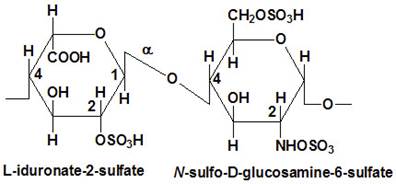

Table showing some G-amino-Gs; http://themedicalbiochemistrypage.org/
Characteristics of GAGsL
|
GAG |
Localization |
Comments |
|
Hyaluronate |
synovial fluid, vitreous humor, |
large polymers, shock absorbing |
|
Chondroitin sulfate |
cartilage, bone, heart valves |
most abundant GAG |
|
Heparan sulfate |
basement membranes, components of cell surfaces |
contains higher acetylated |
|
Heparin |
component of intracellular granules of mast cells |
more sulfated than heparan sulfates |
|
Dermatan sulfate |
skin, blood vessels, heart valves |
|
|
Keratan sulfate |
cornea, bone, cartilage aggregated with chondroitin sulfates |
|
Proteins when they are synthesized, they are modified and transported to their respective destinations. Some proteins while they are threaded into SER and then into Golgi cisternae, they go through modifications, but some modifications are so extensive the final product is huge in size, but very important. One such modified class of proteins is called Proteoglycans.
Proteoglycans
are a class of glycosylated proteins which have covalently linked sulfated glycosaminoglycans,
(i.e., chondroitin sulfate, dermatan sulfate, heparan sulfate, heparin, keratan
sulfate). The protein component of proteoglycans is a core protein which
directs the biosynthesis of proteoglycans to different molecular constructions
and functions. So far, more than 20 genetically different species of core
proteins have been identified. Glycosaminoglycans are sulfated polysaccharides
made of repeating disaccharides (typically a repeat of 40-100 times), which
consist of uronic acid (or galactose) and hexosamines. They endow proteoglycans
with unique functions. The components of disaccharides are (glucuronic
acid/iduronic acid -N-acetylgalactosamine) in chondroitin sulfate and dermatan
sulfate,(glucuronic acid/iduronic acid -N-acetyl glucosamine) in heparan
sulfate and heparin, and (galactose -N-acetyl glucosamine) in keratan sulfate.
Sulfation of glycosaminoglycans in chondroitin sulfate chains is usually
regular, one sulfate per disaccharide throughout the chain, while in heparan
sulfate chains, sulfation is somewhat irregular, and resulting in intensely
sulfated and sparsely sulfated regions on a single glycosaminoglycan chain. In
addition to glycosaminoglycans, proteoglycans normally have other carbohydrate
units including O-linked and N-linked oligosaccharides, as found in other
glycosylate proteins.
Biological functions of proteoglycans derive primarily from those of the
glycosaminoglycan and protein component of the molecule. Glycosaminoglycans
assume extended structures in aqueous solutions because of their strong
hydrophilic nature based on their extensive sulfation, which is further
exaggerated when they are covalently linked to core proteins (a typical example
is aggrecan, see below). They hold a large number of water molecules in their
molecular domain and occupy enormous hydrodynamic space in solution. Another
remarkable property of the glycosaminoglycans, which is particularly
significant in heparan sulfate and heparin, is their capability to specifically
interact with a number of important growth factors and functional proteins.
Such interactions are often crucial to the biological functions of these
proteins. This subject has been drawing much attention and will be further
elaborated in other articles in this series. Properties of core proteins are
also exerted through interactions with other molecules, especially when
proteoglycans are involved in the formation of extracellular matrix.
The
most studied, prototypical proteoglycan is the aggrecan (Figure, panel a),
which constitutes the major component in articular cartilage and accounts for
by far the largest mass among proteoglycans in animal bodies. Aggrecan has a
hyaluronan-binding domain near its amino terminal and forms an enormous
supramolecular structure together with hyaluronan and another protein (called
the "link protein") in articular cartilage tissues. It provides
articular cartilage with its major function of resisting compressional forces.
This was the first proteoglycan molecule found in the late 19th century, was
the main subject of proteoglycan research until the 1970's, and has provided
fundamental knowledge about the molecular structures of proteoglycans.
The biosynthesis of proteoglycans, in addition to the ordinary biosynthetic
processes of O- and N-linked oligosaccharide components as in other
glycoproteins, further requires biosynthesis of glycosaminoglycans in the Golgi
apparatus, which can be considered to be a most complex biosynthetic process
for complex carbohydrates. It requires a number of glycosyl transferases and
sulfo-transferases and involves multiple sub regions of the Golgi apparatus,
which poses a great challenge for researchers in elucidating the organization
and regulatory mechanisms involved in proteoglycan biosynthesis.
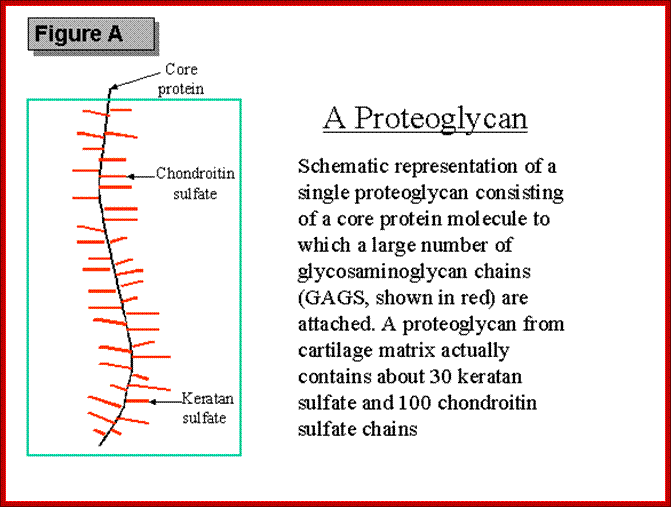
http://webanatomy.net/

Hyaluron chain to which Core proteins are linked by Link proteins, core proteins are added with GAGs. www.suggest-keywords.com
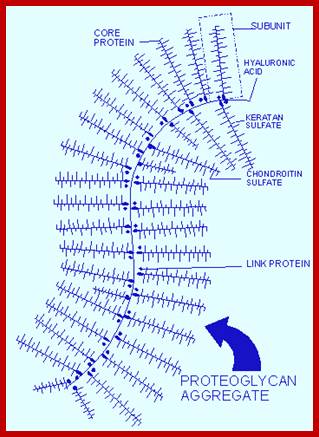
http://jonlieffmd.com
http://webanatomy.net/
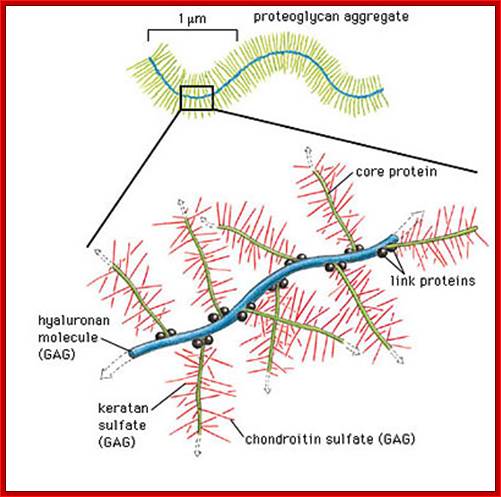
Agrecan: http//:www.studyblue.com
What makes up Aggrecan aggregate?
hyaluronic acid makes up the core, but isn't covalently linked. Aggrecan linked to HA via link protein. Forms brush structure. Type of proteoglycan dominant in cartilage
Fixed charge density in cartilage is provided by: Chondroitin and keratan sulfate
Proteoglycan Proteoglycans consist of a core protein and one or more covalently attached glycosaminoglycan chains Proteoglycans attract water because: Donnan osmotic pressure: caused by the interstitial counterions (for example, Ca2+, Na+) that neutralize the charges on the pro- teoglycans Electrostatic repulsive forces that are developed between the fixed negative charges along the proteoglycan molecules; entrop- ic tendency of the proteoglycan to gain volume in solution.
A diagrammatic view of a proteoglycan aggregate, a part of it is expanded to show hyaluron backbone to which core proteins are linked by link proteins. The core proteins in turn are linked by GAGs.;
http://www.studyblue.com/notes/note/n/physiology
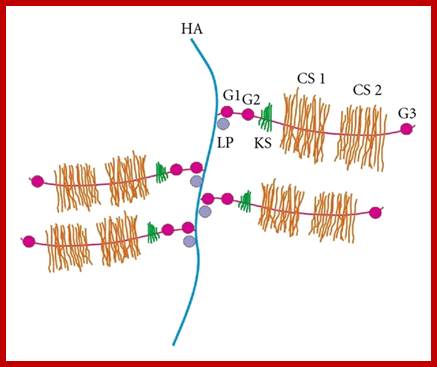
Generalized PG with Hyaluronan chains: Linker proteins. Core protein and GAGs on core proteins.https://www.hindawi.com; http://www.hindawi.com/
|
Some Proteoglycans of Known Sequence |
|
|||
|
Proteoglycan |
Glycosaminoglycan |
Protein |
Number of |
|
|
Secreted or extracellular |
||||
|
Large aggregating |
CS/KS* |
220,952 |
2124 |
|
|
Versican |
CS/DS |
265,048 |
2409 |
|
|
Decorin |
CS/DS |
38,000 |
329 |
|
|
Intracellular granule |
||||
|
Serglycin (PG19) |
CS/DS |
10,190 |
104 |
|
|
Membrane-intercalated |
||||
|
Syndecan |
HS/CS |
38,868 |
311 |
|
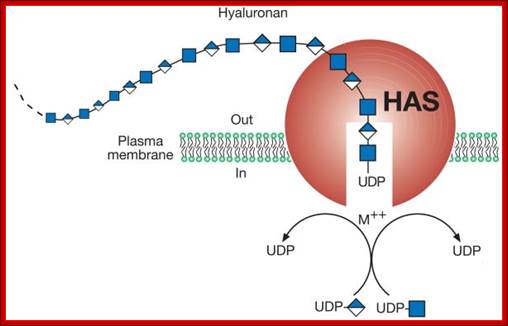
Hyluronan


Structure of the link module: (a) TSG-6 contains a prototypical link fold defined by two α helices (α1 and α2) and two triple-stranded antiparallel β sheets (β1,2,6 and β3–5). (b) The hyaluronan-bound conformation of the protein, with a view showing key amino acids and the position of hyaluronan. (c) A model of a link protein/hyaluronan complex. Binding of the hyaluronan to the protein is mediated through ionic interactions between positively charged amino acid residues (green) and the carboxylate groups of the uronic acids, and by a combination of aromatic ring stacking and H-bond interactions between the polysaccharide and aromatic amino acids (red). In addition, hydrophobic pockets (on either side of Tyr59) can accommodate the methyl groups of two N-acetyl glucosamine side chains, which is likely to be a major determinant in specificity of link modules for hyaluronan.
The structural domains of link protein: Link protein is depicted with three disulphide-bonded domains responsible for the interaction with aggrecan (A) or hyaluronan (B1 and B2). The intact link protein may possess two (LP1) or one (LP2) N-linked oligosaccharides in its amino terminal region. Proteolytic cleavage may occur within the amino terminal region to yield a truncated link protein (LP3). Cleavage may also occur within the A domain to yield fragmented LP.

The structural domains of link protein: Link protein is depicted with three disulphide-bonded domains responsible for the interaction with aggrecan (A) or hyaluronan (B1 and B2). The intact link protein may possess two (LP1) or one (LP2) N-linked oligosaccharides in its amino terminal region. Proteolytic cleavage may occur within the amino terminal region to yield a truncated link protein (LP3). Cleavage may also occur within the A domain to yield fragmented LP.
The central component of the cartilage proteoglycan aggregate is a long molecule of hyaluronan. Bound to it, tightly but noncovalently at 40-nm intervals, are aggrecan core proteins decorated with GAGs). The aggrecan core protein (≈250,000 MW) has one N-terminal globular domain that binds with high affinity to a decasaccharide sequence in hyaluronan; this binding is facilitated by a link protein that binds to the aggrecan core protein and hyaluronan). Covalently attached to each aggrecan core protein, via the trisaccharide linker, are multiple chains of chondroitin sulfate and keratan sulfate. The molecular weight of an aggrecan monomer — that is, the core protein plus the bound glycosaminoglycans — averages 2×106. The entire proteoglycan aggregate, which may contain upward of 100 aggrecan monomers, has a molecular weight in excess of 2×108.
Not all extracellular proteoglycans form large aggregates like aggrecan. A class of proteoglycans present in the basal lamina, for example, consists of a core protein (20,000 – 40,000 MW) to which are attached several heparan sulfate chains. Such proteoglycans bind to type IV collagen and other structural proteins discussed later, thereby imparting structure to the basal lamina.
The aggrecan core protein is depicted with three disulphide bonded globular domains (G1-3), an inter globular domain (IGD), and attachment regions for keratan sulphate (KS) and chondroitin sulphate (CS1 and CS2). The G1 domain is composed of three functional subdomains responsible for the interaction with link protein (A) or hyaluronan (B1 and B2). The CS1 domain exhibits length polymorphism due to a variable number of 19 amino acid tandem repeats. Each repeat contains two serine residues (S) that can act as attachment sites for chondroitin sulphate.
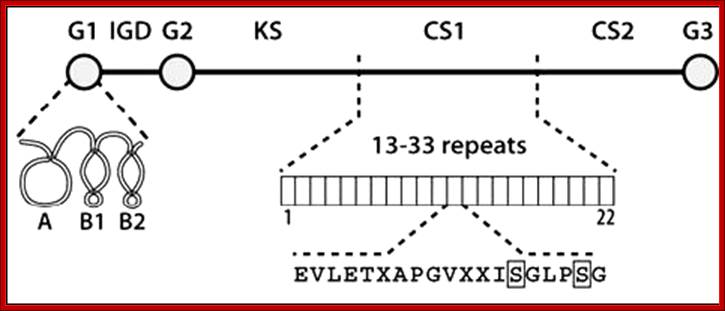
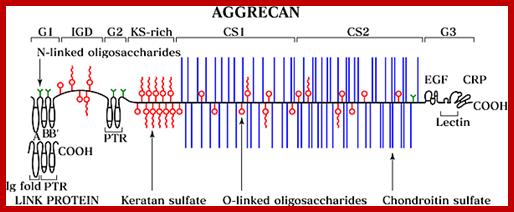
http://glycoforum.gr.jp
The structural domains of aggrecan: The aggrecan core protein is depicted with three disulphide bonded globular domains (G1-3), an inter globular domain (IGD), and attachment regions for keratan sulphate (KS) and chondroitin sulphate (CS1 and CS2). The G1 domain is composed of three functional subdomains responsible for the interaction with link protein (A) or hyaluronan (B1 and B2). The CS1 domain exhibits length polymorphism due to a variable number of 19 amino acid tandem repeats. Each repeat contains two serine residues (S) that can act as attachment sites for chondroitin sulphate.
The schematic representation below each model shows the organization of peptide domains in aggrecan and PG-M/versican. HABR: hyaluronic acid-binding region; GAG: glycosaminoglycan-attachment domain; EGF: epidermal growth factor-like repeat; LEC: C-type lectin-like module; C: complement regulatory protein-like module.
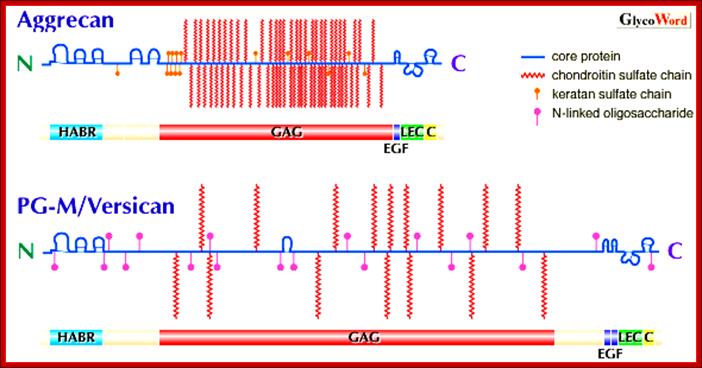
Core protein its N terminal attached to poly-hyaluron chain is studded with GAGs of different kinds starting from the N-terminal region of the protein. CS = Chondroitin sulphate, KS= Keratan sulphate, http://www.glycoforum.gr.jp/
- Proteoglycans have a polysaccharide chain of Hyaluronate as the main thread or primary chain. Hyaluronate chain is made up of disaccharide units of Glucuronic acid or Iduronic acid linked to N-acetyl glucosamine or N-acetyl galacatosamine. The length of this chain can be any 50 000 pairs or more.
This diagram shows different kinds of proteoglycans on the surface of the membrane.
Then the primary chain is added with a number of polypeptide chains at intervals as branches; the protein chain can be 300-400 amino acids long. Protein chains are held to poly-Hyaluronate chains with linker proteins. The protein chain acts as core for the addition of oligosaccharide chains again at intervals.
Mushroom Proteoglycans where the core protein is linear and decorated with branched GAGs
- The oligosaccharide chains are made up of disaccharide subunits such as chondroitin sulphate, heparan sulfate, dermatan sulphate and keratan sulphate. Disaccharide GAGs are added to core proteins’ serine with a sequence motif of, Gly-X-Gly-Ser, where serine residue is added with O-linked glycosylation; the linkage to protein is---Ser--Xyl—GAL--GAL-[Glu-Glu]2-[Glu-Glu]2-[Glu-Glu]n.
- The length of oligosaccharide chains added can vary from 50 to 100 or more. Even the number of disaccharide units per oligosaccharide chain can be 20 to 50 or more. The kind of oligo chains added to the protein from the base to the tip can also vary.
|
Proteoglycan |
Location |
Core Mr (KD) |
Composition |
|
Agrecan, Versican |
ECM |
225-250 |
HA, CS, KS |
|
Decorin, Biglycan-fibronodulin |
Collagen associated |
40 |
CS, DS, KS |
|
G.syndecan, Glypican, Betaglycan, Cerebroglycan, CD44 |
Cell surface |
33,60,92 |
HS, CS |
CS = Chondroitin SO4.
DS = Dermatan SO4.
KS = Keratan SO4.
HA = Hyaluronate.
HS = Heparan SO4
This figure depicts modification of core protein that starts in ER continues in Golgi and completes; then they are sorted and packaged into specific vesicles and transported.
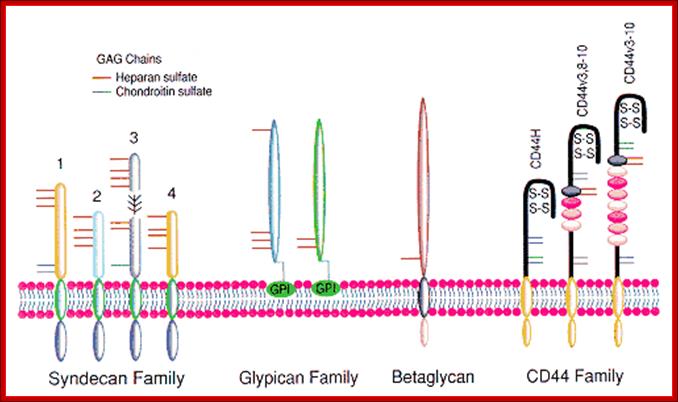
https://www.researchgate.net
Family of proteoglycans like Syndecan, Glypican, Betaglycan and CD44, which vary in their anchoring to the membrane, and the kind of GAGs organized on protein core.
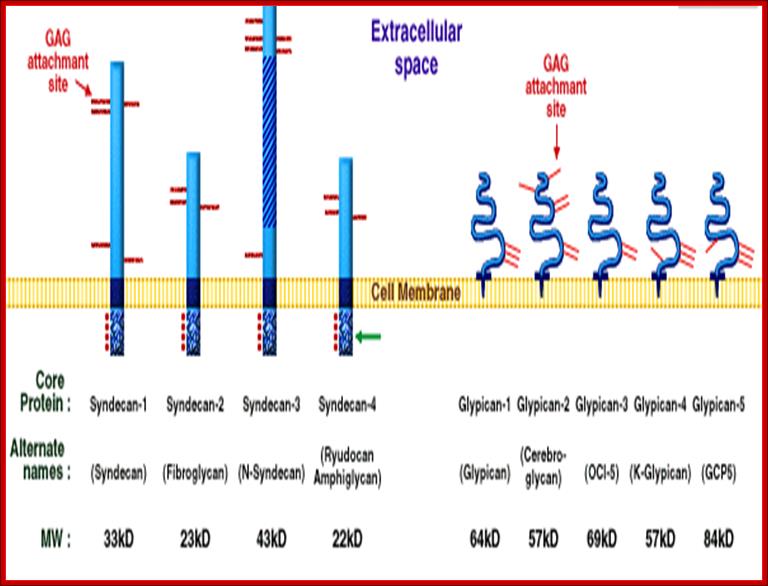
A diagram showing different types of Syndecans and Glypicans anchored to membranes. http://www.glycoforum.gr.jp
Molecular organization of small leucine-rich proteoglycans:
The core proteins don’t represent their actual size, neither the illustrated sites of glycosylation and LRR represented.
![]()
http://www.glycoforum.gr.jp
Some of the simple Proteoglycans as shown above and below are independent molecule but anchored in the membranes; example Perlecan, Syndecan , Bamacan and Agrecan.
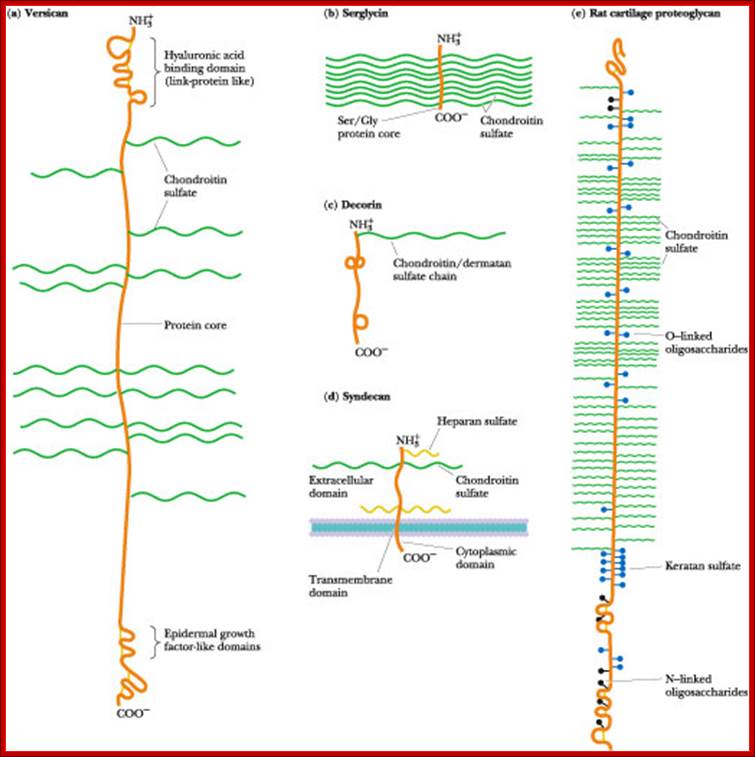
Types of Proteoglycans-Vericilan, seriglycin, syndecan, cartilage PG
Perlecan:
Perlecan is a
major Heparin sulfate (HSPG) molecule of the basement membrane. However, it is
also expressed in embryonic interstitial tissues and stromata in pathologic
conditions. The core protein of perlecan, 467 KDa (human), is composed of five
domains. The amino-terminal domain I contains three heparan sulfate (HS)
chains; domain II has four LDL receptor-like repeats; domain III contains
repeats resembling those in the short arm of laminin; domain IV has Ig-like
repeats similar to those in NCAM, and domain V contains repeats similar to
those in laminin A chain and EGF motifs. Such a domain structure suggests that
the core protein itself carries multifunctional properties, although there is
little actual evidence for them. Its HS chains are heterogeneous in size;
however, each HS chain averaged 380 KDa in molecular mass and 87 nm in length
when examined by electron microscopy. A hybrid variant of perlecan with
dermatan sulfate chains has been reported. Like other HSPG molecules, various
functions of HS chains including binding with several growth factors are being
revealed. In addition to HS chains, perlecan bears a total 20 KDa of N- and
O-linked oligosaccharide chains, which suggested to function in the perlican
secretion.

http://www.glycoforum.gr.jp/
Syndecan: Its molecular weight is about 50000 KD. It has single membrane pass and its carboxyl end is towards cytoplasm, but the N-terminal region is in the exoplasm. The extra cellular domain contains three Heparan sulfate GAGs and two chondroitin sulfate GAGs. It binds to a variety of ligand and modulates receptor signal transduction events.
Aggrecan: Its mol. weight is approximately 250000 kd. It has multiple chondroitin sulfate and keratan sulfate GAGs protein disaccharides bound to serine residues with trisaccharide linkage. Hundreds of decorated proteins bind to single Hyaluronate polymer, producing a proteoglycan aggregate of 2x10^8 Daltons. Its associated water content is equal to the volume of a bacterial cell. Agrin was found as a protein which induces clustering of acetylcholine receptors and acetylcholine esterase at the postsynaptic membrane of neuromuscular junctions. Recently, it has been shown to be a HSPG and distributed in basement membranes other than synapses. In the GBM, agrin is much more abundant than perlecan. The core protein of agrin, 250 KDa (chicken), is composed of the amino-terminal globular head containing laminin binding sites, a second rod-like domain resembling follistatin, and a third globular domain containing laminin G. It is predicted that it has four each of HS chains and N-linked oligosaccharide chains. The HS chains have been shown to bind to NCAM in nervous tissues. Agrin isoforms localized in synaptic sites are products of special alternative splicing.
It is one of the major proteoglycans in cartilage, it has 2316 aa.
The protein component of proteoglycans is synthesized by ribosomes and translocated into the lumen of the rough endoplasmic reticulum. Glycosylation of the proteoglycan occurs in the Golgi apparatus in multiple enzymatic steps. First a special link tetra saccharide is attached to a serine side chain on the core protein to serve as a primer for polysaccharide growth. Then sugars are added one at a time by glycosyl transferase. The completed proteoglycan is then exported in secretory vesicles to the extracellular matrix of the cell. Most important pr xx ns, but disaccharide glycan also very important structural and functional components
Bamacan:
Bamacan, a chondroitin sulfate (CS) proteoglycan of the basement membrane, is localized in various kinds of basement membranes except for the glomerular capillary loop. The core protein of Bamacan, 138 kDa (mouse), consists of five domains: the amino-terminal globular head, the second rod attached with one each of CS chain and a N-linked oligosaccharide chain, the third globular, the fourth rod containing two N-linked oligosaccharide chains, and the fifth globular tail with two CS chains. The function of Bamacan has not been well elucidated yet.
Agrin:
Agrin was found as a protein which induces clustering of acetylcholine
receptors and acetylcholine esterase at the postsynaptic membrane of
neuromuscular junctions. Recently, it has been shown to be a HSPG and
distributed in basement membranes other than synapses. In the GBM, agrin is
much more abundant than perlecan. The core protein of agrin, 250 KDa (chicken),
is composed of the amino-terminal globular head containing laminin binding
sites, a second rod-like domain resembling follistatin, and a third globular
domain containing laminin G. It is predicted that it has four each of HS chains
and N-linked oligosaccharide chains. The HS chains have been shown to bind to
NCAM in nervous tissues. Agrin isoforms localized in synaptic sites are
products of special alternative splicing.
Glypican:
![]()

Schematic of glypican protein structure at the cell surface. The protein is held in the plasma membrane by a GPI anchor at the carboxyl terminus. Numerous glycosoaminoglycan (GAG) attachment sites close to the membrane surface allow heparin and chondroitin sulphate chains to be attached to the core protein (shown in green). The amino terminal end of the protein is a globular structure held together by a conserved set of cysteine residues forming disulphide bridges Glypican: http://atlasgeneticsoncology.org
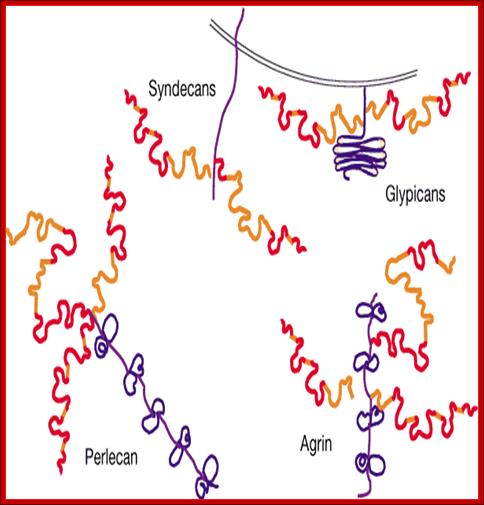
http://www.nature.com
The syndecans and glypican family members are integral membrane proteins. The syndecans core proteins are apparently highly extended transmembrane proteins that contain a short (34–38-residue) carboxy-terminal cytoplasmic domain. The HS chains on the syndecans are linked to serine residues that are distal from the plasma membrane. The glypican core proteins are apparently disulphide-stabilized globular core proteins linked to the plasma membrane by an apparently extended C-terminal region that culminates in a GPI linkage. The HS chains on the glypicans are linked to serine residues adjacent to the plasma membrane in the apparently extended region. Basement membranes in mammalian tissues contain pelican and agrin. Perlican is in nearly all basement membranes as well as in cartilage, whereas agrin is a component of basement membranes in muscle, myoneural junctions, the central nervous system, lung and kidney. Both are large multi domain proteins bearing HS chains near their amino termini. Although both proteins exist as multiple isoforms that arise by alternative splicing and contain globular domains that are homologous to laminin G and epidermal growth factor domains, on the basis of their genomic organization, they are quite distinct proteins. http://www.nature.com

Some of activities involving cell surface heparan sulfate proteoglycans. HSPGs control activity and catabolism of various enzymes (A), participate in cell adhesion (B) and modulate growth factor signalling (C). (A) LPL is one of the molecules, which are internalized while bound to HSPG, and syndecan-1 was identi®ed as one of the proteoglycans responsible for LPL endocytosis. (B) Focal adhesion is a specialized membrane complex in which integrins and HSPGs cooperate to connect extracellular matrix and cytoskeleton. The HSPG involved appears to be exclusively syndecan-4. (C) Binding to HSPGs can regulate growth factor interactions with tyrosine kinase receptors. Several models were proposed to explain the role of HSPGs in FGF-2 signalling. The one depicted here shows HS chain mediating dimerization of FGF-2 and subsequently FGF receptors, promoting formation of a signalling complex and tyrosine kinase signal transduction. Multiple HSPGs were shown to support FGF-2 biological activities. Components are not drawn to scale. https://www.researchgate.net

The six members of the vertebrate glypican family share a characteristic structure. Glypicans are anchored to the cell surface via a GPI linkage, have a conserved pattern of 14 cysteine residues, which contribute to intramolecular disulfide linkages, and display GAG attachment sites predominantly near the membrane. Alternative names and molecular weight of the core proteins are also indicated; https://www.rndsystems.com Glypican variations.

Hutmacher DW, Cool S – ;https://openi.nlm.nih.gov

A structural
model of aggrecan and PG-M/versican.
The schematic representation below each model shows the organization of peptide
domains in aggrecan and PG-M/versican. HABR: hyaluronic acid-binding region;
GAG: glycosaminoglycan-attachment
domain; EGF: epidermal growth factor-like repeat; LEC: C-type lectin-like
module; C: complement regulatory protein-like module.; Toshikazu Yada;http://www.glycoforum.gr.jp
3D structure of collagen; .; http://www.medicalnewstoday.com

Collagen is the most abundant protein in the human body and is the substance that holds the whole body together. It is found in the bones, muscles, skin and tendons, where it forms a scaffold to provide strength and structure; there 16 types of collagens. Each chain contains around 1,000 amino acids, two alpha1 chains one alpha2 chains and usually features an amino acid sequence consisting of glycine, proline and hydroxyproline. http://www.medicalnewstoday.com

Native triple helices are characterized by their resistance to proteases, such as pepsin or trypsin 9 and can only be degraded by specific collagenases (metalloproteinases); https://www.proto-col.com