Co and Post Translational Events.
Bacterial Systems:
And Eukaryotes:
In prokaryotes, transcription and translation are coupled. A large number of the proteins are synthesized on ribosomes bound mRNAs as polysomes. Polyribosome formation enhances the rate of protein synthesis. Are any ribosomes associated with plasma membranes? No, but yes is the answer now. Some of the proteins destined to periplasm or secreted to exoplasm bind to plasma membranes ribosome receptors/ protein translocons. The proteins in the process of translation or translated are destined to different loci such as plasma membranes, exoplasmic space and outer membrane in gram negative bacteria and many remain free in cytoplasm.
In eukaryotic cells, synthesis of proteins takes place, basically at two sites: one in free-state, means, the protein synthesis is often localized in the intracellular space. However the location is predetermined for some. The other site is endoplasmic reticulum. The initiation of protein synthesis starts in free- state, then depending upon the N-terminal sequence they are left to synthesize either in free-state, or bind to ER and as the protein synthesis starts based on the N terminal sequence the ribosome binds to ER and the protein is transferred into ER lumen. Once the proteins are synthesized they are fold3ed and move to ER vesicles and budded off and reach their destination extra cellular and intracellular sites. Can we consider the inner surface of the plasma membrane as another site? No?
Whether protein is synthesized ER bound or free state at any intracellular sites, proteins are targeted to certain destinations such as intracellular organelles such as ER, Plasma membranes, Lysosome, Tonoplast (plants), Chloroplasts (plants), Mitochondria, Golgi complex, Peroxisomes, Nucleus or many can be secretary. The so called free-state in some cases is not actually free, the translating mRNAs are held by Actin filaments or small proteins belong to microtrabaculae (?), but they are localized within the cell. Two other sites of protein synthesis in eukaryotic cells are mitochondria and plastids, which are similar to prokaryotic.
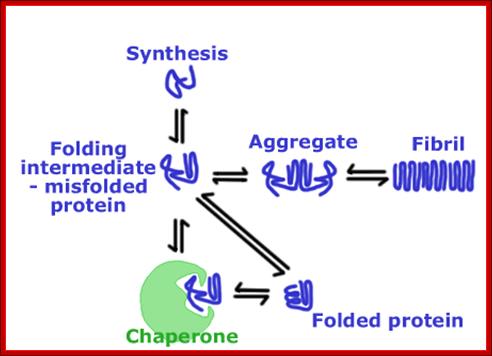
As proteins exit from the large ribosomal exit groove, proteins depending upon their N terminal a.a sequences start organizing into specific structural forms. This folding takes place at rapid pace and automatic. This folding is assisted by binding HSP like (Heat shock Proteins like) chaperones.
Various forces act upon the protein chain to fold in specific ways-they are 1. ionic (lys - asp), 2.hydrophobic and VDW (phe - val), 3. H-bond (ser - ser), 4. H-bond to ionic (asp - asn) 5. Van der Waals (ser -ala). Hydrophobic sequences initiate immediate attracting to each other and collapse with hydrophobic domains in water free region and hydrophilic regions outer such that they interact with water. Among bondings S-S boding provides stability to proteins. If the position of S-S boding not proper, in such cases they are broken and reform in correct positions, thus proteins in structural and functional form work properly; if they are not folded properly, often they aggregate and they are subjected to ubiquitination and degradation by proteasome mode.
Folding of proteins assisted by Chaperones and Chaperonins in Bacteria and Eukaryotes:
Proteins, whatever may be their size, as they emerge out of ribosomal exit channel, they are guided by chaperones and use protein folding machinery and get folded into 3-D structural form. The class-I of chaperones are found to be heat shock proteins expressed constitutively and also in response to heat shock/stress; and assist nascent proteins to fold and unfolded proteins to refolded into their original functional state.

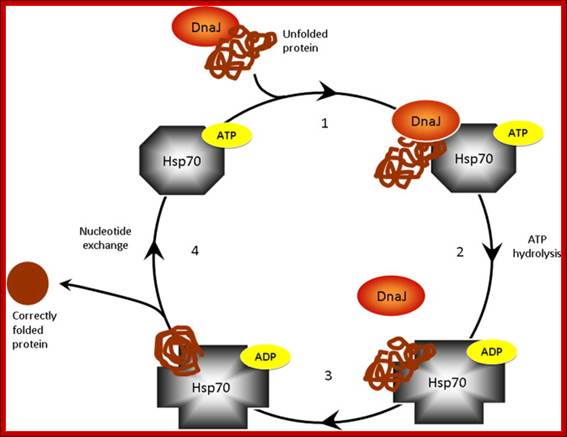
Initiatiall DnaK and DnaJ are involved in guiding the protein chain to fold; if it fails proteins are guided to chaperonins for folding; Macmillan Publishers Ltd: Nature 475, 324-332 (2011) doi:10.1038/nature10317;
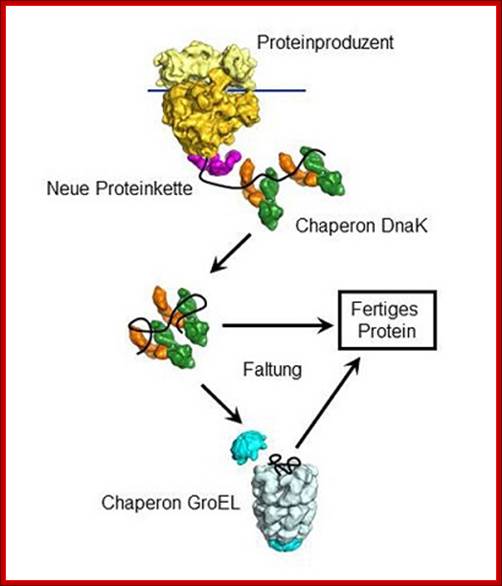
The chaperone DnaK
binds to new proteins and mediates their folding. Proteins if they cannot fold DnaK
transports to GroE, chaperonin-a molecular chaperone, a highly specialized
folding machine. © MPI of Biochemistry.
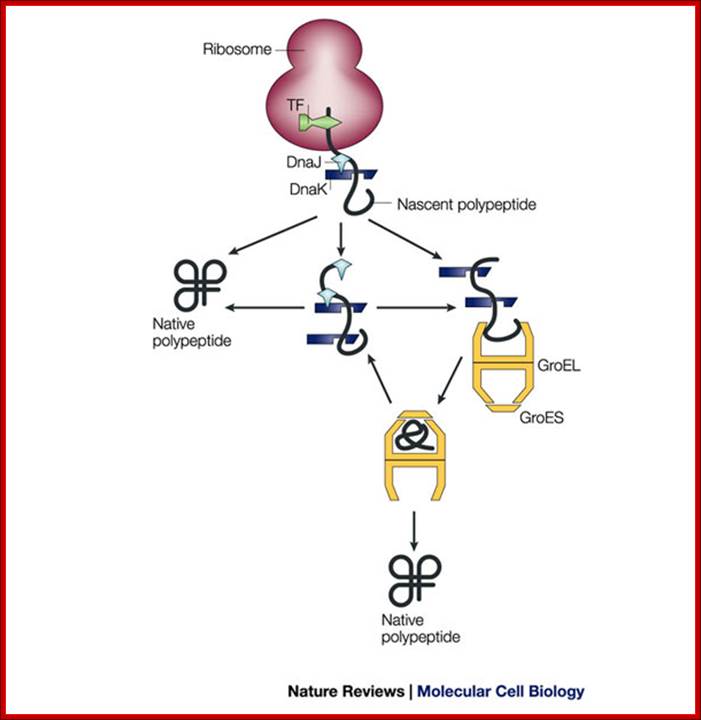
Pathways of chaperone mediated folding;
The general mechanism by which chaperones carry out their functions usually involves multiple rounds of regulated binding and release of an unstable conformer of target polypeptides. The four main chaperone systems in the Escherichia coli cytoplasm are as follows. (1) Ribosome-associated trigger factor that assists in the folding of newly-synthesized nascent chains. (2) The Hsp 70 system consisting of DnaK (Hsp 70), its cofactor DnaJ (Hsp 40), and the nucleotide exchange factor GrpE. This system recognizes polypeptide chains in an extended conformation. (3) The Hsp 60 system, consisting of GroEL (Hsp 60) and its cofactor GroES (Hsp 10), which assists in the folding of compact folding intermediates that expose hydrophobic surfaces. (4) The Clp ATPases which are typically members of the Hsp 100 family of heat shock proteins. These ATPases can unfold proteins and disaggregate preformed protein aggregates to target them for degradation. Several advances have recently been made in characterizing the structure and function of all of these chaperone systems. These advances have provided us with a better understanding of the protein folding process in the cell; Houry W.A.
The first set of proteins studied, many years ago, were involved in protein folding, they were called chaperone; they were found to assist the assembly of nucleosomes from folded histones and linear DNA. Chaperones bind to nascent polypeptide chains and also partially folded proteins and prevent them from aggregating and misfolding.
There is another class of chaperones called chaperonins which are involved in folding and unfolding of proteins, often called molecular chaperones e.x GroEL/GroES in E.coli called group I chaperonins. Group II chaperonins are found in eukaryotes, e.x TRiC (TCP-1 ring complex).
Surfactants as Protein Chaperones:
The University of Chicago has developed novel intellectual property around the use of biocompatible surfactants to assist proper folding and refolding of proteins which has been licensed to Maroon. The mechanism by which surfactant chaperones do this is illustrated below. When a protein is denatured, it becomes stabilized in a new conformation by its interaction with the water molecules surrounding the protein. When certain classes of surfactants are added, their hydrophobic sections (blue segment of surfactant copolymers illustrated below) are attracted to hydrophobic regions of the protein. This puts highly polar parts of the surfactants (pink) into the water surrounding the protein, allowing the surfactants to disrupt the water structure, lower surface tension and facilitate refolding. While there are many folding agents that are effective, multiblock copolymer chaperones are effective at concentrations well tolerated by the body.
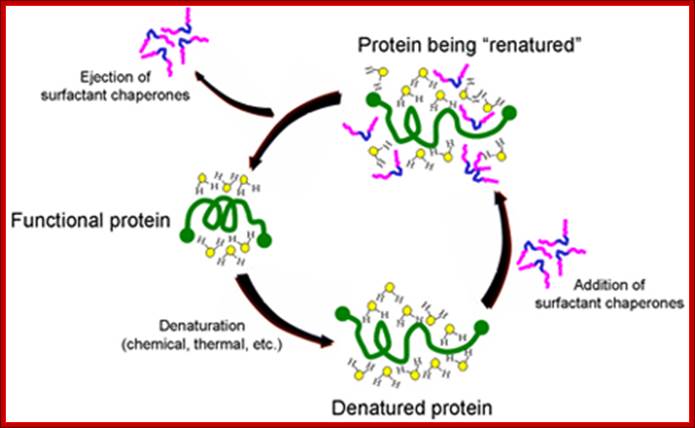
Maroon's scientists, with an advanced understanding of the pharmacology of such copolymer chaperones, are developing formulations of a combination therapy with complementary compounds that will allow synthetic chaperones to be used at concentrations below those which cause undesired side effects. This "cocktail" will enable Maroon Biotech to administer a therapy that is safe in the body. The Maroon development team is also using its knowledge of surfactant chaperones to explore the possibility of inventing new, more effective compounds. http://www.maroonbiotech.com
Chaperone Assisted Protein folding:
Heat shock proteins are the most abundant class of proteins expressed in cells. They are the members of the heat shock protein family, which is upregulated in response to stress. Hsp90 is found in bacteria and all branches of eukarya, but it is apparently absent in archaea. They play an important role in protein-protein interactions such as folding and assisting in the establishment of proper protein conformation (shape) and prevention of unwanted protein aggregation. By helping to stabilize partially unfolded proteins, HSPs also aid in transporting proteins across membranes within the cell. Heat-shock proteins also occur under non-stressful conditions, simply "monitoring" the cell's proteins. There are several families of chaperones; those most involved in protein folding are the 40-kDa heat shock protein (HSP40; DnaJ), 60-kDa heat shock protein (HSP60, GroEL), and 70-kDa heat shock protein (HSP70; DnaK) families.

Effects of Nascent Chain Binding Chaperones on the Folding of Multidomain Proteins, a Working Model: The translating polypeptide chain of a hypothetical two-domain protein is shown in pink with folded domains represented by hexagons and squares. Bacterial chaperones are in blue, and eukaryotic chaperones are in green. See Discussion for details. Vishwas R Agashe et al: http://www.cell.com/
The chaperone network of the prokaryotic cytosol:
Jason C. Young, Vishwas R. Agashe, Katja Siegers and F. Ulrich Hartl
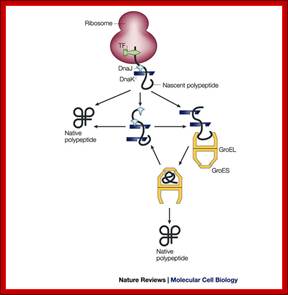
Chaperone-assisted protein folding in E.coli is further aided by GroEL/GroES chaperonin complex; http://www.nature.com/
Nascent polypeptide chains are met by trigger factor (TF) ribosome associated chaperone, as they emerge from the ribosome exit tunnel. The 70-kDa heat-shock protein DnaK, which is stimulated by its J-domain co-chaperone DnaJ, also binds nascent polypeptides. Newly synthesized polypeptides can fold spontaneously or can be assisted by DnaK. Alternatively, they can be passed on to the GroEL–GroES chaperonin system for final folding, and, in some cases, might again interact with DnaK. Folding is ATP dependent.
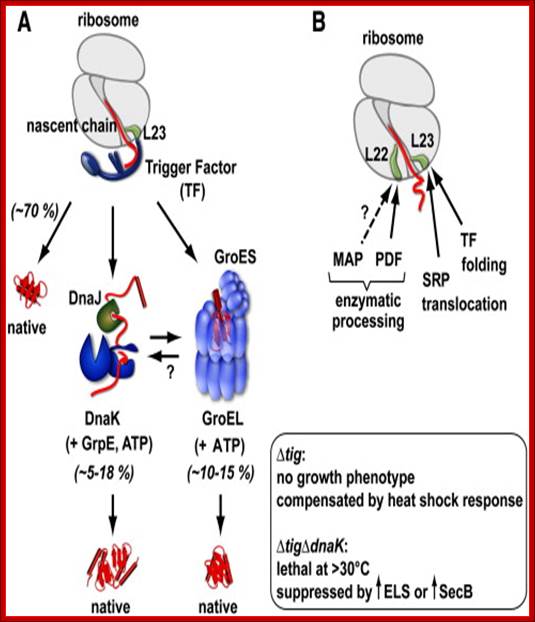
Trigger Factor function and interplay with other factors. (A) Model of chaperone action in folding of newly synthesized cytosolic proteins in E. coli. Nascent polypeptides initially interact with ribosome-bound Trigger Factor (TF). Upon release from TF, they either fold spontaneously (roughly estimated two thirds of cytosolic proteins under normal growth conditions) or require further folding-assistance by downstream chaperones, namely the Hsp70 chaperone DnaK, which acts together with its co-chaperone DnaJ and the nucleotide exchange factor GrpE (KJE system), and/or the Hsp60 chaperone GroEL with its co-chaperone GroES (ELS system). The ATP-dependent DnaK- and GroEL-machineries may act co- and/or post-translationally (see Section 10 for details). (Inset) The lack of TF is compensated by induction of the cellular heat shock response with enhanced KJE and ELS action. The absence of both TF and DnaK is synthetically lethal at temperatures above ∼ 30 °C, but can be partially suppressed by overexpression of the secretion-dedicated chaperone SecB or the ELS system. (B) Ribosome-associated factors acting on nascent chains. The ribosomal protein L23 constitutes the major binding site for both TF and the signal recognition particle (SRP), which targets membrane proteins for co-translational translocation. TF and SRP were shown to co-exist at the ribosome where they likely compete for nascent chains. Peptide deformylase (PDF) and methionine aminopeptidase (MAP) enzymatically process the N-termini of nascent proteins. PDF binds to a groove in between ribosomal proteins L22 and L32 with major contacts to L22. The possibility that MAP may bind to ribosomes is indicated, however such an interaction of bacterial MAP with ribosomes has not been demonstrated to date. Study of influence of co-translational protein folding within the ribosome tunnel on trigger factor recruitment; Huang, Joseph Jen-Tse; http://www.chem.sinica.edu.tw/
Trigger Factor (TF) represents the only
ribosome-associated chaperone known in bacteria. It is found in bacteria and
chloroplasts, whereas structurally different ribosome-associated factors exist
in the archaeal and eukaryotic cytosol.
Despite of its absence in the eukaryotic cytosol, TF shares a certain
functional similarity with eukaryotic ribosome-associated chaperones. When
expressed in S. cerevisiae, E. coli TF
binds to yeast ribosomes and partially complements the knockout phenotype of
the yeast ribosomal chaperone triad consisting of Ssb, Ssz and Zuotin. E. coli TF
is a constitutively expressed and abundant cytosolic protein that exists in a
two- to three-fold molar excess relative to ribosomes (∼ 50 µM
TF versus ∼ 20 µM ribosomes). It transiently associates with
ribosomes in a 1:1 stoichiometry using ribosomal protein L23 as major docking
site; ·
Anja
Hoffmann, · Bernd
Bukau![]() ,
, ![]() , · Günter
Kramer; BBA 2010.
, · Günter
Kramer; BBA 2010.

Suthep Wiyakrutta; Mahidol University;http://www.sc.mahidol.ac.th/
Models for the chaperone-assisted folding of newly synthesized
polypeptides in the cytosol.
( A ) Eubacteria. TF, trigger factor; N, native protein. Nascent chains
probably interact generally with TF, and most small proteins ( ~ 65 to 80% of
total) fold rapidly upon synthesis without further assistance. Longer chains
(10 to 20% of total) interact subsequently with DnaK and DnaJ and fold upon one
or several cycles of ATP-dependent binding and release. About 10 to 15% of
chains transit the chaperonin system?GroEL and GroES?for folding. GroEL does
not bind to nascent chains and is thus likely to receive an appreciable
fraction of its substrates after their interaction with DnaK. ( B ) Archaea.
PFD, prefoldin; NAC, nascent chain-associated complex. Only some archaeal
species contain DnaK/DnaJ. The existence of a ribosome-bound NAC homolog, as
well as the interaction of PFD with nascent chains, has not yet been confirmed
experimentally. ( C ) Eukarya-the example of the mammalian cytosol. Like TF,
NAC probably interacts generally with nascent chains. The majority of small
chains may fold upon ribosome release without further assistance. About 15 to
20% of chains reach their native states in a reaction assisted by Hsp70 and
Hsp40, and a fraction of these must be transferred to Hsp90 for folding. About
10% of chains are co- or posttranslationally passed on to the chaperonin TRiC
in a reaction mediated by PFD (Hartl and Hayer-Hartl 2002).

http://www.sc.mahidol.ac.th/
The nascent chain is stabilized in a folding-competent state during translation by the Hsp70 chaperone system (DnaK, DnaJ) (1 and 2). These chaperones bind hydrophobic segments exposed by the extended chain that will later be buried within the folded structure. Upon completion of translation, the protein is unable to fold using the Hsp70 chaperone system and must be transferred into the central cavity of GroEL. This step requires GrpE, the nucleotide exchange factor of DnaK (3). After binding of the protein in a molten globule–like conformation into the open ring of GroEL (4), the protein is encapsulated by GroES in the folding cage (5). Folded protein emerges from the cage as GroES unbinds (6). The model was later extended to include the cooperation of DnaK with the ribosome-bound chaperone trigger factor and the finding that the Hsp70 system mediates the folding of proteins that do not require the physical environment of the chaperonin cage; http://www.nature.com/
Chaperone-assisted protein folding in E.coli cytosol, the path of chaperone assisted proteins with the help of GroEL and with ATP energy are loaded into chaperonins for proper folding and release. F Ulrich Hartl; http://www.nature.com/
The nascent chain is stabilized in a folding-competent state during translation by the Hsp70 chaperone system (DnaK, DnaJ) (1 and 2). These chaperones bind hydrophobic segments exposed by the extended chain that will later be buried within the folded structure. Upon completion of translation, the protein is unable to fold using the Hsp70 chaperone system and must be transferred into the central cavity of GroEL. This step requires GrpE (EK), the nucleotide exchange factor of DnaK (3). After binding of the protein in a molten globule–like conformation into the open ring of GroEL (4), the protein is encapsulated by GroES in the folding cage (5). Folded protein emerges from the cage as GroES unbinds (6). The model was later extended to include the cooperation of DnaK with the ribosome-bound chaperone trigger factor and the finding that the Hsp70 system mediates the folding of proteins that do not require the physical environment of the chaperonin cage, F Ulrich Hartl, Nature Medicine 2011.
A list of Chaperones:
- Chaperone (protein), a protein that assists the non-covalent folding/unfolding in molecular biology
- Co-chaperone, a protein that assists a chaperone in protein folding and other functions.
Proposed model of chaperone-assisted protein folding by the HSP70–DNAJ complex; Although HSP70 also functions in the regulation of protein transport, degradation and protein–protein interactions, one of the best described roles for the HSP70 chaperone is in the folding of nascent or denatured polypeptides, in cycles that are controlled by ATP and the association with DNAJ co-chaperones. (1) DNAJ forms a transient association with the unfolded protein (substrate) via exposed hydrophobic residues for delivery of the substrate to HSP70. (2) DNAJ binds to the ATP-bound form of HSP70, and transfers the misfolded or unfolded protein to the substrate binding cleft of HSP70. DNAJ stimulates the hydrolysis of ATP to ADP by the ATPase domain of HSP70. The ADP-bound form of HSP70 has a higher affinity for the protein substrate than the ATP bound form, and thus binds tightly to the unfolded protein and DNAJ leaves the complex. (3) Unfolded protein is prevented from aggregation or non-productive folding pathways. (4) The ATP-bound form of HSP70 is regenerated by the activity of nucleotide exchange factors (such as BAG1). ATP bound Hsp70/DnaK has a lower affinity for the substrate and so the protein is released, to fold to its normal conformation or re-associate with another chaperone. The chaperones and co-chaperones are now able to continue the chaperone-assisted protein folding cycle ; Jason N. Sterrenberg, Gregory L. Blatch, Adrienne L. Edkins
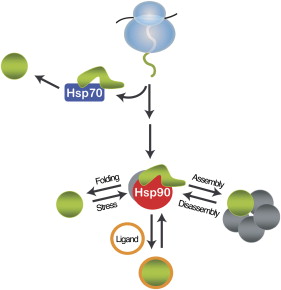
A comprehensive understanding of the cellular functions of the Hsp90 molecular chaperone has remained elusive. Although Hsp90 is essential, highly abundant under normal conditions, and further induced by environmental stress, only a limited number of Hsp90 "clients" have been identified. To define Hsp90 function, a panel of genome-wide chemical-genetic screens in Saccharomyces cerevisiae were combined with bioinformatic analyses. This approach identified several unanticipated functions of Hsp90 under normal conditions and in response to stress. Under normal growth conditions, Hsp90 plays a major role in various aspects of the secretory pathway and cellular transport; during environmental stress, Hsp90 is required for the cell cycle, meiosis, and cytokinesis. Importantly, biochemical and cell biological analyses validated several of these Hsp90-dependent functions, highlighting the potential of our integrated global approach to uncover chaperone functions in the cell; McClellan AJ, Xia Y, Deutschbauer AM, Davis RW, Gerstein M, Frydman J
Chaperone are also called by names such as Foldases which help in protein folding, ex.GroEL/GroES; unfoldases- to unfold proteins. Another class called Holdases- help and prevent aggregation- ex.DnaJ or Hsp33. Chaperones not just restricted to fold /assist folding of proteins, they are also involved in binding DNA and various types of RNA molecules to properly fold into their functional shape, they are called RNA chaperones; Daniel Herschlag.
RNA Chaperones:
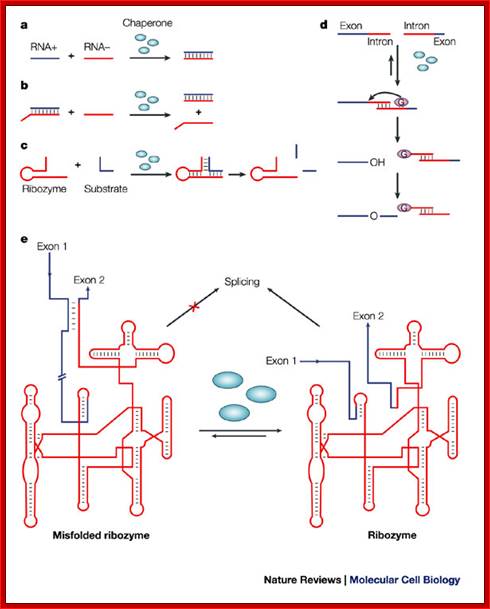
RNA-chaperone activity was monitored by using different in vitro and in vivo assays. Annealing of two complementary RNA oligonucleotides (RNA+ and RNA-) is accelerated in the presence of proteins with RNA-chaperone activity (blue ellipses)142 (see figure part a). Proteins with RNA-chaperone activity accelerate the displacement of a homologous RNA oligonucleotide from a duplex RNA in the strand-displacement assay143 (see figure part b). Substrate binding and product dissociation are accelerated in the cleavage reaction of the hammerhead ribozyme62 (see figure part c). The group-I intron of the thymidylate synthase (td) gene from phage T4 was split into two halves. Assembly of the two halves requires heating to 55°C, and splicing therefore does not take place, and is only detectable in the presence of proteins with RNA-chaperone activity at 37°C (Ref. 77) (see figure part d). Proteins with RNA-chaperone activity rescue splicing of the td group-I intron in vivo by rescuing structures that are trapped in non-native states55 (see figure part e). The pink ellipse that contains a G represents the guanosine cofactor, which is covalently attached to the intron during the splicing reaction, and is the hallmark of group-I introns; Renée Schroeder, Andrea Barta and Katharina Semrad;; http://www.nature.com/
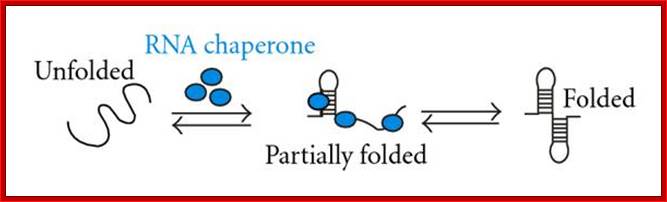
Hypothetical mechanisms of RNA chaperoning. (a) shows folding of an RNA molecule in the presence of RNA chaperones (blue). RNA chaperones and proteins with RNA chaperone activity prevent the RNA from misfolding and increase annealing of the correct structure by crowding. (b) Proteins with RNA chaperone activity possessing disordered regions (blue) interact with misfolded RNA. Upon energy transfer, the RNA structure loosens and the disordered protein domain becomes more ordered. Proteins with RNA chaperone activity are dispensable in both cases after the RNA has folded into its native form; http://www.hindawi.com
The principal heat-shock proteins that have chaperone activity belong to five conserved classes: HSP33, HSP60, HSP70, HSP90,HSP100, and the small heat-shock proteins (sHSPs).
|
Approximate molecular weight (kDa) |
Prokaryotic proteins |
Eukaryotic proteins |
Function |
|
GroES |
Hsp10 |
||
|
20-30 kDa |
GrpE |
The HspB group of Hsp. Eleven members in mammals includingHsp27, HSPB6 or HspB1 [28] |
|
|
DnaJ |
Hsp40 |
Co-factor of Hsp70 |
|
|
GroEL, 60kDa antigen |
Hsp60 |
Involved in protein folding after its post-translational import to the mitochondrion/chloroplast |
|
|
DnaK |
The HspA group of Hsp including Hsp71, Hsp70, Hsp72, Grp78 (BiP), Hsx70 found only in primates |
Protein folding and unfolding, provides thermotolerance to cell on exposure to heat stress. Also prevents protein folding during post-translational import into the mitochondria/chloroplast. |
|
|
HtpG, C62.5 |
The HspC group of Hsp including Hsp90, Grp94 |
Maintenance of steroid receptors and transcription factors |
|
|
100 kDa |
ClpB, ClpA, ClpX |
Hsp104, Hsp110 |
Tolerance of extreme temperature |
The table is not complete?!
ER- resident Chaperones:
In the endoplasmic reticulum (ER) there are general, lectin and non-classical molecular chaperones helping to fold proteins.
· General chaperones: GRP78/BiP, GRP94, GRP170.
· Lectin chaperones: calnexin and calreticulin
· Non-classical molecular chaperones: HSP47 and ERp29
· Folding chaperones:
· Protein disulfide isomerase (PDI),
· Peptidyl prolyl cis-trans-isomerase (PPI),
· ERp57
Bacterial:
Hsp60 GroEL/GroES 1MD- two rings of heptamer (seven) each. GroES heptamer acrs as a cover to barrel. Hsp70 Dnak, Hsp40 DnaJ, Hsp90-(HtpG) even found in EuK, require co-chaperones,
HSP100-form hexameric: In the presence of ATP. These proteins are thought to function as chaperones by processively threading client proteins through a small 20 Å (2 nm) pore, thereby giving each client protein a second chance to fold.
A list of chaperones:
Hsp90α,
Hsp90β,
TRAP1
HtpG
Hsp70,
Hsp40 90 C,
ER Dimer, α-helix/β-sheet layers 65
Hsp70,
Hsc70,
BiP MHsp70;,
DnaK Hip, Hop,
GrpE 70 C,
M, ER N- and C-terminal domains 9
Hsp40 DnaJ
Hsp70,
Hsc73 40 C, M,
ER Bi-helical 5
Small Hsps: IbpA, IbpB, Hsp70 - 12–43 C Oligomers; α-Crystallin domain 32.
ER Chaperones- Calnexin and Calreticulin;
Difference between Chaperones and Chaperonins:
Chaperonins are a
subtype of molecular chaperones. So, Chaperonins ‘are’ chaperones--but not all
chaperones are chaperonins. It's like the "squares are rectangles but not
all rectangles are squares". In this case chaperonins are squares, and
chaperones are "rectangles" which include both squares and other
types of rectangles. Chaperonins are subgroup of chaperones; it is large
family of proteins.
Group I Chaperonins:
Group I chaperonins are found in bacteria as well as organelles of endosymbiotic origin: chloroplasts and mitochondria. Hsp60–Hsp10e GroEL–GroESe, Gim C 57 M, Two heptameric rings; homo-oligomeric. They are all called molecular chaperones.
- GroEL is a double-ring 14mer , two rings of seven each, with a greasy hydrophobic patch at its opening and can accommodate the native folding of substrates 15-60 kDa in size.
- GroES is a single-ring heptamer that binds to GroEL in the presence of ATP or transition state analogues of ATP hydrolysis, such as ADP-AlF3. It's like a cover that covers GroEL (box/bottle).
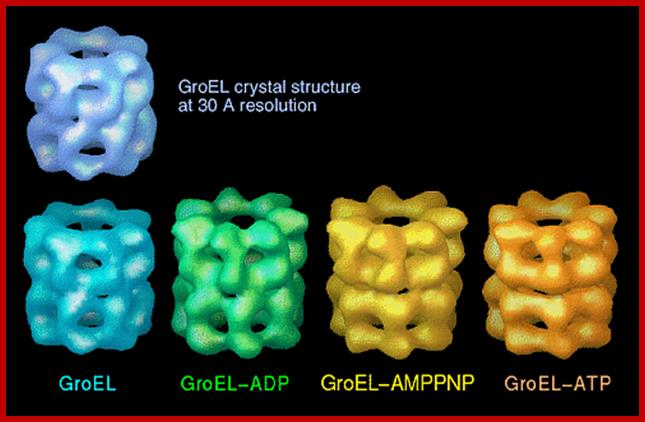
GroEL-Nucleotide structure at 30A resolution; http://people.cryst.bbk.ac.uk/~ubcg16z/cpn/elseries.gif
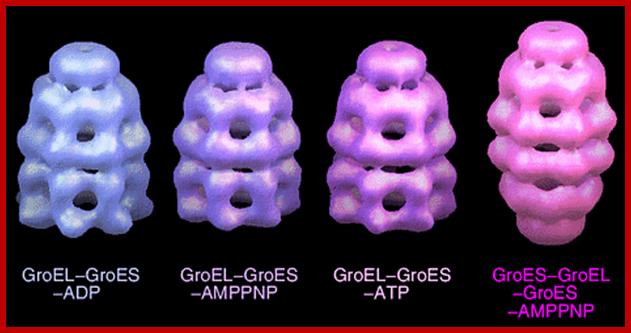
GroEL-GroES-nucleotide structures at 30 Å resolution ; GroEL consists of two rings of Octamers and GroES consists of One ring of heptamer on the top of GroEL.Helen Saibil, Lunchun Wang et al; http://people.cryst.bbk.ac.uk/
Gr oES and GroEL side
view, two barrels of seven subunits in each ring ;show four banded appearance
mol.wt of them is 60kDa; landry@mailhost.tcs.tulane.edu; http://www.tulane.edu/
oES and GroEL side
view, two barrels of seven subunits in each ring ;show four banded appearance
mol.wt of them is 60kDa; landry@mailhost.tcs.tulane.edu; http://www.tulane.edu/

GroES. "Top view" C-alpha trace colored by crystallographic B-factors. Only one of seven mobile loops is visible at left. It is made up of seven identical subunits of 10kDaeach. For a closer look examine the kinemage estour_2.kin. [If necessary, first get the kinemage viewer Mage 4.2; http://www.tulane.edu/

Group I chaperonins; http://pdslab.biochem.iisc.ernet.in/
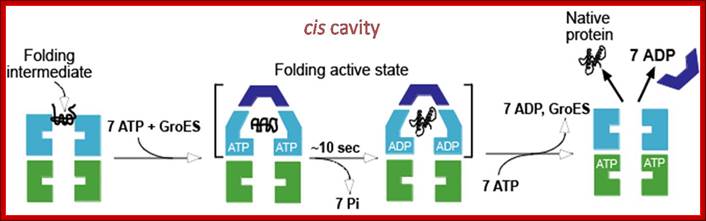
· 1: The reaction cycle of the bacterial GroEL/GroES chaperonin system. The molecules are shown in cross-sections. Each GroEL subunit consists of a central domain an apical substrate-binding domain and a connecting intermediate region. The binding of unfolded substrate protein to one GroEL ring is followed by binding of ATP and the co Chaperonins GroES. The substrate is released into the now-closed ring cavity where it can fold. Following ATP hydrolysis, the binding of ATP to the opposite ring triggers the dissociation of GroES and the dissociation of folded substrate protein from the complex.
- 2: Model for the reaction cycle of the archaeal Mm-cpn chaperonin system. The molecules are shown in cross-sections. Each Mm-cpn subunit consists of a central domain (red) an apical substrate-binding domain (dark green) and a connecting intermediate region (yellow). The binding of unfolded substrate protein (light blue) to one Mm-cpn ring is followed by binding of ATP. This event induces conformational changes in helical domain protrusions (light green), leading to closure of the cylindrical cavity. The substrate folds inside the chaperonin complex and dissociates from it after ATP hydrolysis has occurred.
Chaperonins:
The bacterial chaperonin GroEL consists of two homo-heptameric rings, which form a barrel-shaped structure. Each subunit has an equatorial ATPase domain where the rings contact each other, and an apical substrate-binding domain at the open end of the barrel. Alternating ATP cycles in each ring control the conformations of the apical domains. The GroES co-chaperone is a homo-heptameric structure that can also bind to the GroEL apical domains. After substrate binding to one ring of GroEL (see figure, part a), ATP and GroES are bound by the same ring, which displaces the substrate into an enclosed cavity that is capped by GroES (part b). Sequestration of the substrate (part c) allows folding without the risk of non-productive intermolecular contacts, which could otherwise lead to aggregation. Dissociation of GroES after ATP turnover allows substrate release (part d)19, 20. Some polypeptides require several cycles of binding and release to reach their native state. Certain polypeptides that are too large to fit inside the chaperonin cavity can also be bound by the ring of GroEL that is opposite the ring that is bound by GroES, and their folding can be assisted through cycles of such trans binding of GroES.
GroES.
The eukaryotic chaperonin TCP1 ring complex (TRiC) is structurally related to GroEL, but has greater similarity to the chaperonins of archaea (thermosomes). TRiC has two rings of eight different subunits, which also have equatorial ATPase domains and apical substrate-binding domains49. Structural similarity to thermosomes indicates that extensions of the TRiC apical domains mediate the encapsulation of a substrate without the need for a GroES-like co-chaperone51, 52. Similar to GroEL, ATP-dependent movements of the apical domains are involved in TRiC-mediated folding50, as they bind substrate polypeptides and release them into the enclosed central cavity53. TRiC has no known co-chaperones that modulate its activity. Mammalian prefoldin binds TRiC with a comparatively weak affinity66, but a regulatory function has not been observed. An open question is how important each of the eight TRiC subunits are for the folding of different proteins.
The chaperonins ; Pathways of chaperone-mediated protein folding in the cytosol http://www.nature.com/
(a) Non-native protein binds to the trans ring of a GroEL–GroES complex. (b) End-to-end exchange of GroES (possibly through a symmetric complex with a GroES molecule bound to each of the GroEL rings) results in the encapsulation of the protein substrate in the cis cavity. (c) In the presence of ATP, productive folding of protein substrates can occur in the cis cavity. (d) Release of GroES and substrate protein (whether folded or not) results in the regeneration of the acceptor complex for non-native protein. Note that for simplicity, changes in the GroEL–nucleotide state have been omitted. Neil A. RANSON1, Helen E. WHITE and Helen R. SAIBIL Department of Crystallography, Birkbeck College London, Malet Street, London WC1E 7HX, U.K.
Mechanism of Escherichia coli GroEL-GroES: A vertical section of the GroEL double-ring is shown representing the three-domain structure of the GroEL subunits (Reprinted from Cell 125, Tang et al., Structural Features of the GroEL-GroES Nano-cage Required for Rapid Folding of Encapsulated Protein (2006), with permission from Elsevier)- Chaperonin-assisted Protein Folding; Manajit Hayer-Hartl
Group II Chaperonins:
Group II chaperonins, found in the eukaryotic cytosol and in archaea, are more poorly characterized. TRiC (TCP-1 Ring Complex, also called CCT for chaperonin containing TCP-1), is the eukaryotic chaperonin, It is composed of eight different though related subunits; each thought to be represented once per eight-membered ring. TRiC was originally thought to fold only the cytoskeleton proteins actin and tubulin but is now known to fold dozens of substrates. TCP-1, or CCT TF55,56 , thermosome, 55 C N Octameric/nonameric rings;
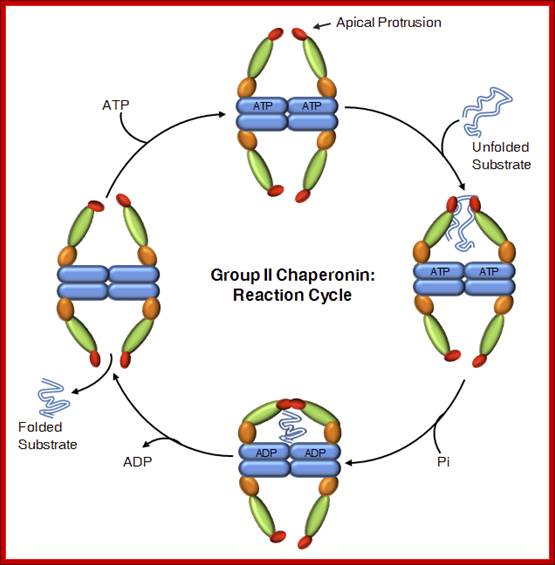
Reaction Cycle; Group II chaperonins
In contrast to GroEL which is stress induced, TRiC is expressed constitutively and is required for folding of essential proteins. Some of TRiC substrate exceeds 100 kD size, and many of them cannot be folded by classical prokaryotic and eukaryotic chaperones. https://www.google.co.in

https://commons.wikimedia.org
Comparison of the basic conformations of group I (GroEL-GroES) and group II chaperonins within the functional cycle. For the sake of simplicity the schematic drawings of the chaperonin-substrate complexes show only single rings.

These chaperones do not work alone and in many cases they function in a coordinated manner. Another objective is therefore the structural characterization of the interaction between some of these chaperones, in particular the interaction between CCT and other chaperones such as prefoldin (PFD), the phosducin-like protein (PhLP), Hsp70, Hsp110, Hsp40,
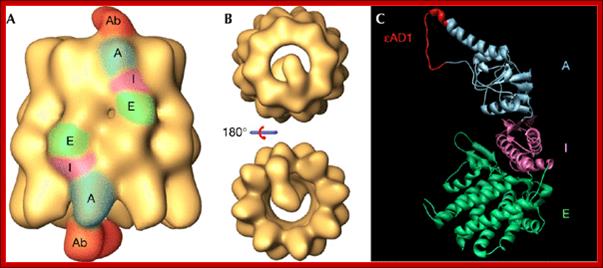
Three‐dimensional reconstruction generated by cryoelectron microscopy of the complex between CCT and the CCTε εAD1 monoclonal antibody. (A,B) Side and end‐on views, respectively, of the CCT–εAD1 complex. In (A), the equatorial (E), intermediate (I) and apical (A) domains, and the part of the antibody reconstructed (Ab) are indicated and coloured. (C) Epitope localisation of εAD1, which interacts with residues Pro260–Tyr274 of CCTε. The binding domain is shown in red in the homologous region of the structure of the α‐thermosome from Thermoplasma acidophilum (Ditzel et al, 1998; PDB 1A6D). As in (A) the regions marked E, I and A correspond, respectively, to the equatorial, intermediate and apical domains. εAD1, epsilon apical domain 1; CCT, chaperonin containing TCP‐1. http://embor.embopress.org/
Thermosomes:
The Thermosome, the Chaperonins of the archaea, and its homologue from the cytosol of eukaryotes, known as TRiC or CCT, form a distinct subfamily of the Chaperonins that does not depend on a co-chaperonin for protein folding activity. Recent structural data obtained by cryo- electron microscopy and X-ray crystallography provide the first insights into a novel mechanism remarkably different from that of the bacterial GroEL-GroES system, Klumpp M. Baumeister W.
We have determined to 2.6 Å resolution the crystal structure of the thermosome, the archaeal group II chaperonin from T. acidophilum. The hexadecameric homolog of the eukaryotic chaperonin CCT/TRiC shows an (αβ)4(αβ)4 subunit assembly. Domain folds are homologous to GroEL but form a novel type of inter-ring contact. The domain arrangement resembles the GroEL-GroES cis-ring. Parts of the apical domains form a lid creating a closed conformation. The lid substitutes for a GroES-like cochaperonin that is absent in the CCT/TRiC system. The central cavity has a polar surface implicated in protein folding. Binding of the transition state analog Mg-ADP-AlF3 suggests that the closed conformation corresponds to the ATP form, Lars Ditzel et al

Three-dimensional map of the thermosome obtained by cryoelectron tomography. The thermosome is the archetype of the group II chaperonins, which, besides the archaeal members, also comprise the chaperonins of the cytosol of eukaryotes, known as TRiC or CCT (for a recent review, see Ref. 40). Thermosomes embedded in ice were reconstructed individually, before the data set was subjected to three-dimensional (3-D) alignment, classification and averaging. The map shown in (a) is calculated from 227 particles; the tilt increments were 6° and the effective tilt range was ±65°. The cumulative dose for recording the whole data set was ∼2000 e-nm−2. The reconstruction was low-pass filtered with a cut-off at (2.8 nm)−2. The thermosome is depicted in the open – i.e. substrate accepting – conformation. A 90° segment was removed to provide a view of the interior of the complex. (b) Tomographic map in a transparent representation with crystal structures of the domains fitted into it. The equatorial domains are shown in yellow, the intermediate domains in blue and the apical domains in orange (base) and green (apical or lid subdomain; for details, see Ref. 41). The open thermosome has a diameter of ∼15 nm and a height of ∼18 nm. By combining the low-resolution tomographic map with the high-resolution domain structures, the resolution gap is bridged and pseudo-atomic structures are obtained. Thus, detailed pictures of molecular assemblies or of conformational states can be obtained that are difficult to capture or preserve in crystallization experiments. Wolfgang Baumeister, Rudo Grimm, and Jochen Walz.
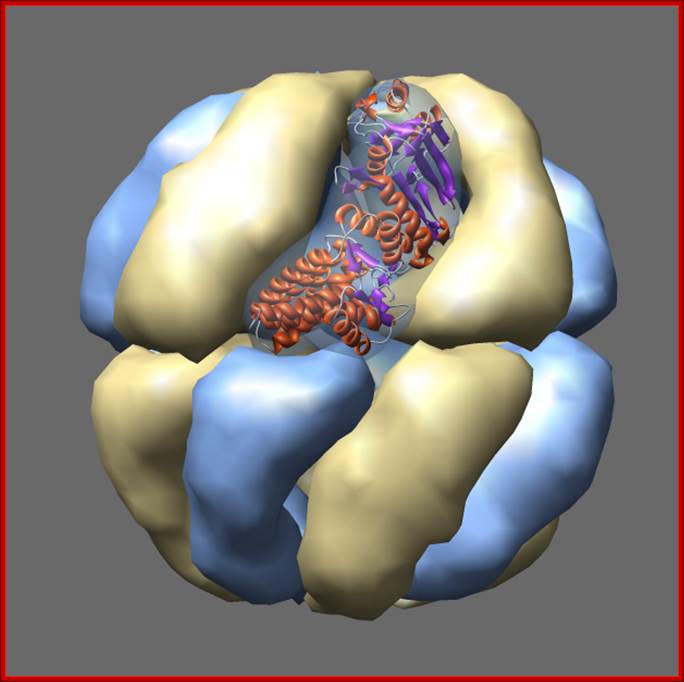
Thermosomes are hollow balls inside which proteins are folded. They are found in the cytosol of eukaryotes and in archaea. Eukaryotic thermosomes have 8 different protein subunits, while archaeal ones are composed of one, two or three different proteins. The one shown from Thermoplasma acidophilum has two distinct proteins colored blue and yellow, each present in 8 copies. The two proteins have 60% sequence identity and are very similar in structure. One monomer is shown as a ribbon. Actin and tubulin are folded by eukaryotic thermosome. Protein Data Bank model 1a6d.
www.bioinfo.imdik.pan.pl/wiki
Alhas crystalline are chaperones, g-crystallins are lens proteins. Although the role of amyloid fibrils in diseases like Alzheimer's disease is unclear, in the case of cataract the most likely role of the aggregates is simply to scatter light, causing lens fogging.
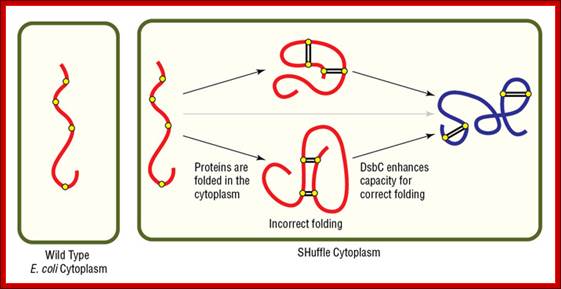
Protein stability by disulphide bond formation:
Role of Glutaredoxin in disulphide bond formation, in E.Coli
https://www.neb.com
Sulfide bond formation and folding go hand in hand. Almost all proteins as they come out of large ribosomal exit tunnel they are assisted by chaperones and prevent them wrongly fold or get aggregated. At the same time wherever the protein chains free from chaperones they naturally assume spontaneous structural folding due hydrophobic regions which collapse as a bundle and hydrophilic regions on the outer side of the collapsed region. For proper folding and stabilization, the presence of Cysteine residues at pivotal sites are recognized and bind by Glutaredoxin (containing glutathione-glu-cys-gly), Thioredoxin, BIP proteins, Protein Disulfide Isomerase PDI components; which facilitate disulfide bond formation thus they bring about proper folding into 3-D structure with proper motifs and domains.
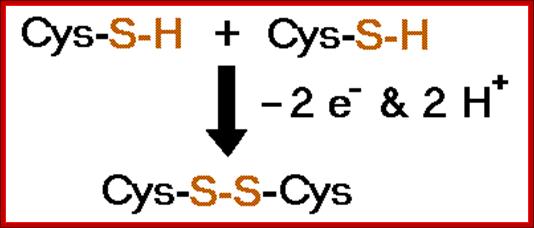
Various forces act upon the protein chain to fold in specific ways-they are 1. ionic (lys - asp), 2.hydrophobic and VDW (phe - val), 3. H-bond (ser - ser), 4. H-bond to ionic (asp - asn) 5. Van der Waals (ser -ala). But there is one strong force that contributes to the folding and stabilization of proteins. That is the disulphide bond (S-S), and it differs from all other bonds in being a covalent bond. It can only be formed between the side chains of two Cysteine residues. The side chain of cysteines-CH2-SH contains a sulfhydryl group -SH. Two sulfhydryl’s can react with oxygen to lose their 2 hydrogen atoms (H with its electron, not H+ ions, not protons) and they covalently bind to each other in the process.
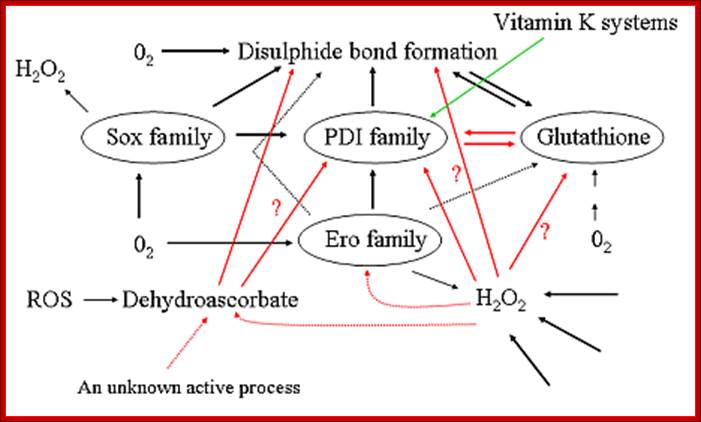
Pathways for disulfide bond formation in the ER. Dotted arrows represent hypothetical pathways, red arrows indicate pathways in which our group has recently generated significant new data, the green arrow indicates a pathway for which the physiological relevance is unclear but which has recently been shown to be the major route in the periplasm of some bacteria. ROS = reactive oxygen species; http://www.oulu.fi/biocenter/groups/ruddock/detailsProject Leader: Prof. Lloyd Ruddock, Ph.D.

from Wikipedia; https://www.michaeljfox.org;http://guweb2.gonzaga.edu/
http://sciencesway.net/
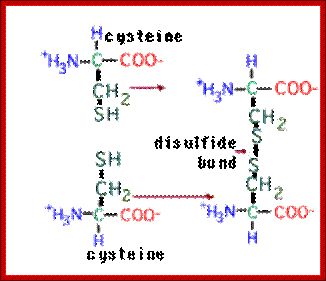
The amino acid cysteine undergoes oxidation and reduction reactions involving the -SH (sulfhydryl group). The oxidation of two sulfhydryl groups results in the formation of a disulfide bond by the removal of two hydrogens.;
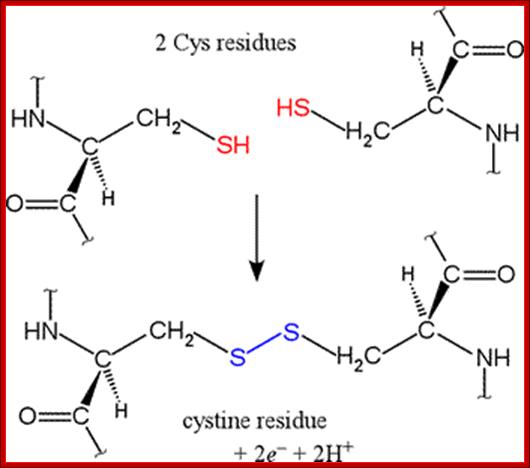
A disufide bond is a covalent chemical bond between two sulfur atoms derived from two sulfhydryl or thiol groups. In biochemistry, these thiol groups are usually from the side chains of the amino acid cysteine. Shown at right are two cysteine residues in polypeptide chain(s). The thiol groups are in their reduced forms (in red in figure). Removal of two hydrogens (H+ + e−) from each thiol (by an oxidizing agent, not included in the figure, which represents an oxidation half-reaction), and concomitant formation of a new covalent bond - the disulfide bond - between the two sulfur atoms yields the lower structure at right. A disulfide-linked pair of cysteine residues is termed a cystine residue. The conversion of two sulfhydryl groups to a disulfide linkage is an oxidation reaction. Conversely, disulfide bonds can be reduced to yield two thiols, which is the reverse of the half-reaction shown at right.
2 Cys + R-S-S-R → cystine + 2 RSH
β-mecaptoethanol (2-mercaptoethanol) and dithiothreitol (DTT) break the sulfhydryl bond reducing disulfide linkages. http://guweb2.gonzaga.edu/
Protein-CH2-SH + HS-CH2-Protein + üO2 ---> Protein-CH2-S-S-CH2-Prot + H2O
SH bond formation between two cysteine residues with the elimination of 2e- and 2H+; http://www.bmb.uga.edu/
Thiol-disulfide chemistry :
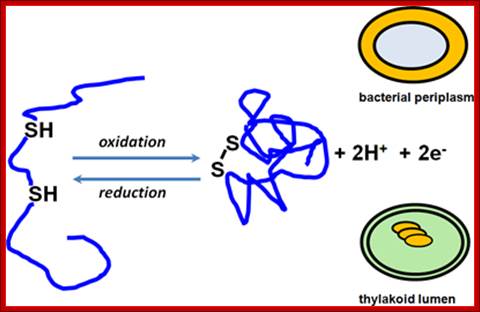
Thiol-disulfide chemistry; Sulfhydryl oxidation leads to disulfide bond formation and
disulfide bond reduction yields free thiols. These reactions are under the
control of catalysts, which were best studied in the bacterial periplasm. Our
group is interested in detailing the operation of such pathways in the
thylakoid lumen, a compartment topologically related to the periplasm.;Thiol-disulfide chemistry ; http://www.biosci.ohio-state.edu/
Sulfhydryl oxidation leads to disulfide bond formation and disulfide bond reduction yields free thiols. These reactions are under the control of catalysts, which were best studied in the bacterial periplasm. Our group is interested in detailing the operation of such pathways in the thylakoid lumen, a compartment topologically related to the periplasm.; Bond formation leads to folding; http://www.biosci.ohio-state.edu/
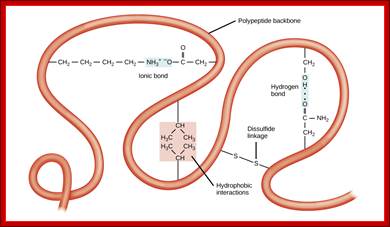
The tertiary structure of proteins is determined by a variety of chemical interactions. These include hydrophobic interactions, ionic bonding, hydrogen bonding and disulfide linkages; http://cnx.org/
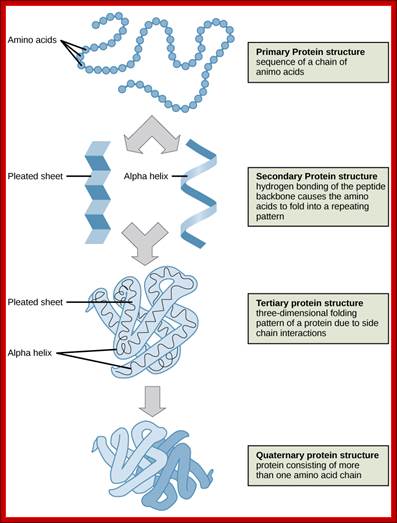
Bottom Fig; The four levels of protein structure can be observed in these illustrations. (credit: modification of work by National Human Genome Research Institute) ; http://cnx.org/

Anfinsen wanted to show that the information for protein folding resided entirely within the amino acid sequence of the protein. He choose ribonuclease A as his model for folding but he couldn't completely denature the protein unless he treated it with the denaturant urea plus 2ME to break the disulfide bridges. Anfinsen discovered that removing 2ME but not urea led to recovery of 1% of the activity. This is attributed to the formation of random disulfide bridges between the 8 cysteines present in the protein. There are 105 different possibilities (7x5x3x1) so the 1% recovery makes sense. It also shows that the correct three-dimensional conformation must be achieved fairly rapidly when urea is removed since most of the protein under those conditions becomes active; http://chemistry.umeche.maine.edu/
Anfinsen made three suggestions:
- The protein itself contains all the information needed to get the fold right; the cellular enviroment is not necessary
- Since the refolding occurs rapidly, in a matter of seconds, the protein does not explore conformation space randomly
- Therefore, folding likely follows a specific pathway
Some problems with the simplistic interpretation of Anfinsen's experiments:
- The unfolded state is likely an ensemble of structures of similar energies
- Folded proteins likewise may be an ensemble of closely related structures
- As demonstrated by ribonuclease, secondary minima may trap incorrectly folded structures
- Proteins may fold from structures that retain some secondary and even tertiary structure
- Significant evidence exists for some folding occurring while the protein is still on the ribosome [Cabrita et al., Curr. Opin. Struct. Biol., 2010, 20, 33]; http://chemistry.umeche.maine.edu/
Sulfhydryl oxidation leads to disulfide bond formation and disulfide bond reduction yields free thiols. These reactions are under the control of catalysts, which were best studied in the bacterial periplasm. Our group is interested in detailing the operation of such pathways in the thylakoid lumen, a compartment topologically related to the periplasm. http://www.blosci ohio state.edu~plant bio
The redox agent that mediates the formation and degradation of disulfide bridges in most proteins is glutathione, a versatile coenzyme that we have talked before in a different context. Recall that the important functional group in glutathione is the thiol, highlighted in blue in the figure below. In its reduced (free thiol) form, glutathione is abbreviated a GSH
Most proteins fold into a roughly globular shape (enzymes, Hb, antibodies--see a picture of the enzyme lysozyme: a space-filling model, or showing just the backbone connections, or a ribbon model), but many take on an elongated or even fibrous shape (collagen, myosin [in muscle], fibroin [in silk]); (Tim Soderberg University of Minnesota, Morris).
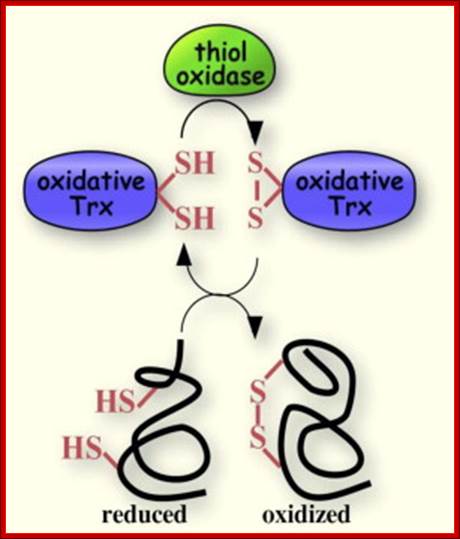
A disulfide forming system through catalyzed reaction in chloroplasts: The thiol oxidase generates disulfides de novo. An oxidative type thioredoxin accepts the disulfide and transfers it to a substrate protein. An isomerase activity, which is coupled in eukaryotes to the oxidative thioredoxin, is required for substrate proteins with of multiple disulfides, Gal Wittenberg Avihai Danon. Mechanism of disulfide reduction by thioredoxin. During the thiol-disulfide exchange reaction, a transient mixed disulfide bond is formed between thioredoxin and its substrate. After completion of the catalytic cycle, a reduced substrate protein is released and thioredoxin is converted to the oxidized form. Trx: thioredoxin; red: reduced; ox: oxidized, Yoshiyuki Matsuoa, b, Junji Yodoia
![]()
![]()
3D Print of Hemoglobin Crystel; http://www.dailybits.com/
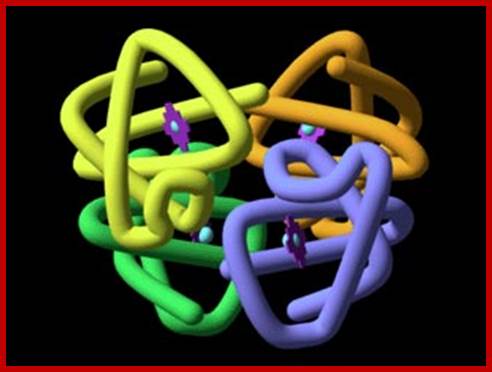
Quarternary protein structure, 3-D assembly of subunits; Hemoglobin; The quarternary structure of hemoglobin comprises two alpha and two beta subunits. The spatial arrangement of each alpha-beta pair is symmetrical. Contacts between the subunits are by means of H-bonds. The heme groupsare inorganic iron (Fe++) groups that assist in binding oxygen (O2): they are not a typical part of the quaternary structure of proteins;. http://home.comcast.net/; https://www.mun.ca/
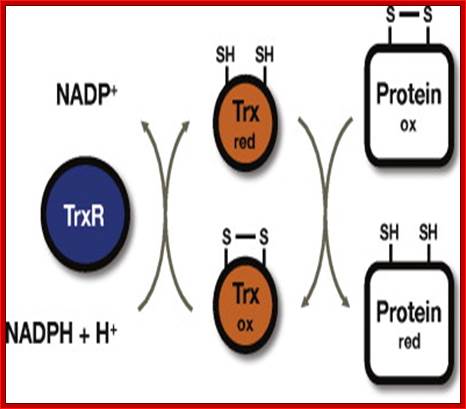
In the bacterial periplasm, a system for the efficient introduction as well as isomerization of disulfide bonds is in place. In eukaryotes, disulfide bonds are introduced into proteins in the endoplasmic reticulum. Both eukaryotic and prokaryotic organisms keep their cytoplasm reduced state and, consequently, disulfide bond formation is impaired in this sub cellular compartment. Disulfide bridges can stabilize protein structure and they are often present in high abundance in secreted proteins. In eukaryotic cells such bonds are formed in the oxidizing environment of endoplasmic reticulum during the export process. Bacteria do not possess a similar specialized sub cellular compartment, but they have both export systems and enzymatic activities aimed at the formation and at the quality control of disulfide bonds in the oxidizing periplasm. Chaperones binding to hydrophobic regions of polypeptide chains facilitate S-S bond formation in the presence of Glutaredoxin Grx. Chaperonins perform refolding of proteins through reformation of S-S bond formation.

Glutaredoxin. http://en.wikipedia.org/
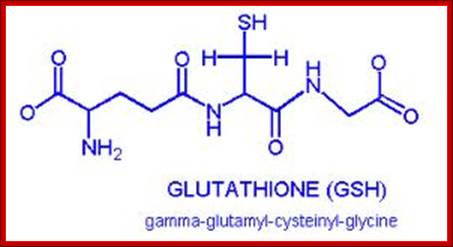
Glutaredoxin are small redox enzymes of 106a.a (Hu) that use Glutathione as cofactor. They are reduced by the oxidation of glutathione; Gln-Cysteine-Glycine (GSH). Oxidized Glutathione is regenerated by glutathione reductase. Grx1 is a cytosolic disulfide oxidoreductase, while Grx2 is found in mitochondria and the nucleus.
http://genomics.unl.edu
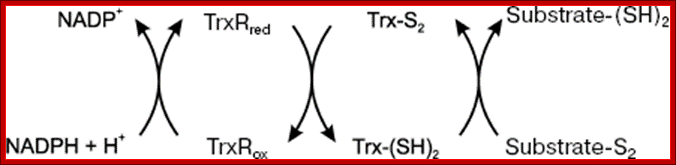
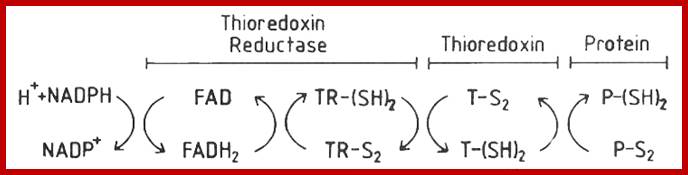
Thioredoxin (Trx) is a small protein contains an active site that involves residue called cysteine; cysteine-X-X-cysteine. Thioredoxin is a hydrogen donor for ribonucleotide reductase. Its reduction process requires NADPH. Both Glutaredoxin and Thioredoxin work together in E.coli as well as in plants.
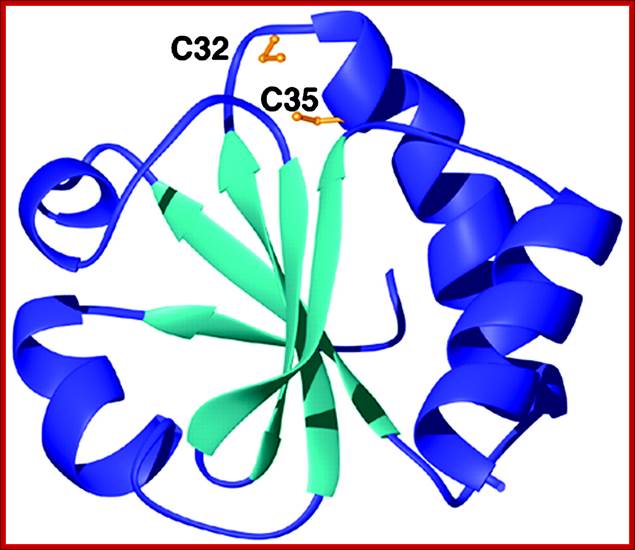
Thioredoxin protein; NMR structure of E. coli thioredoxin. The active site cysteines (C32 and C35) are indicated in yellow. http://www.pnas.org/
Though
these S-S bonds are weak bonds (2.05 °A in length, 60kcal/mole), weaker than
C-C and C-H bonds), but in the aggregate,
they are strong enough to hold the polypeptide fold together at least under the
thermal motion conditions of physiological temperature (37 ° C).
The following diagrams show the formation of S-S bonds among the Cysteine residues in proper orientation and position. If S-S bonds are in wrong position they removed and reformed correctly.
The cytoplasm, of Escherichia coli is constantly maintained in reducing state, this allows thousands of specific reactions. The cytoplasmic reduction of disulfide bonds is achieved by the thioredoxin super family which includes the Glutaredoxin proteins along with glutathione. Members of this protein family are present in most, if not all, living organisms. Glutaredoxin catalyze reversible oxidation/reduction of protein disulfide groups and glutathione-containing mixed disulfide groups. They are involved in different cellular processes such as DNA replication, sulfate assimilation or response to oxidative stress.
http://quizlet.com/
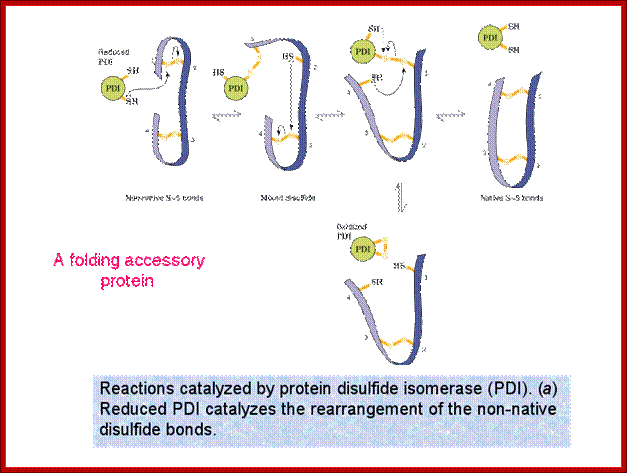
Role of Protein Disuphate Isomerase (PDI); they can change disulfide bond position correct position.
The variety of cellular functions performed by the Glutaredoxin family is made possible, in part, by the wide range of redox potentials associated with their active site Cys-X-X-Cys motif. Glutaredoxin 1 and Glutaredoxin 3 exhibit 33% sequence identity and are structurally nearly superimposable. Yet, Glutaredoxin 1 can reduce the essential enzyme ribonucleotide reductase, which provides deoxy- ribonucleotides for DNA replication, and PAPS reductase, required for cysteine biosynthesis, while Glutaredoxin 3 cannot. These two proteins provide a good model system for understanding the specificity of these proteins.
DsbA-DsbB Interaction through Their Active Site Cysteines: Satoshi Kishigami(1), Eiko Kanaya, Masakazu Kikuchi, and Koreaki Ito.
Formation
of disulfide bonds in Escherichia
coli envelope proteins is facilitated by the Dsb (Disulfide Bond
protein family) system, which is thought to consist of at least two components,
a periplasmic soluble enzyme (DsbA) and a membrane-bound factor (DsbB). DsbA
contain CxxC active center in thioredoxin fold. Although it is believed that
DsbA directly oxidizes substrate cysteines and DsbB reoxidizes DsbA to allow
multiple rounds of reactions, direct evidence for the DsbA-DsbB interaction has
been lacking. We examined intracellular activities of mutant forms of DsbA,
DsbA30S and DsbA33S, in which one of its active site cysteines (Cys![]() or Cys
or Cys![]() , respectively) has been replaced by serine. The DsbA33S protein
was found to dominantly interfere with the disulfide bond formation and to form
intermolecular disulfide bonds with numerous other proteins when cells were
grown in media containing low molecular weight disulfides such as GSSG. In the
absence of added GSSG, DsbA33S protein remained specifically disulfide-bonded
with DsbB. These in
vivo results not only confirm the previous findings that Cys
, respectively) has been replaced by serine. The DsbA33S protein
was found to dominantly interfere with the disulfide bond formation and to form
intermolecular disulfide bonds with numerous other proteins when cells were
grown in media containing low molecular weight disulfides such as GSSG. In the
absence of added GSSG, DsbA33S protein remained specifically disulfide-bonded
with DsbB. These in
vivo results not only confirm the previous findings that Cys![]() of DsbA is hyper-reactive in
vitro but provide evidence that DsbA indeed interacts selectively
with DsbB. We propose that the Cys
of DsbA is hyper-reactive in
vitro but provide evidence that DsbA indeed interacts selectively
with DsbB. We propose that the Cys![]() -mediated DsbA-DsbB complex represents an intermediate state of
DsbA-DsbB recycling reaction that has been fixed because of the absence of Cys
-mediated DsbA-DsbB complex represents an intermediate state of
DsbA-DsbB recycling reaction that has been fixed because of the absence of Cys![]() on DsbA.
on DsbA.
A number of secreted and membrane proteins in both bacteria and eukaryotes contain disulfide bonds. In 1991, evidences were found that bacteria require the periplasmic protein DsbA for efficient formation of such bonds. DsbA is maintained with its active site cysteines in the oxidized state by the membrane protein DsbB. Genetic analysis of DsbA and DsbB shows a structure-function correlation. The isomerization of incorrectly formed disulfide bonds depends on the periplasmic DsbC protein. This protein, which is maintained with its active site cysteines in the reduced state, receives its electrons from thioredoxin in the cytoplasm via a membrane protein DsbD. This is an interesting mechanism of electron transfer across the cytoplasmic membrane.
Exploring to understand the role of the various disulfide reducing components of the cytoplasm is going on. These include the Thioredoxin and Glutaredoxin, but alternative pathways for disulfide bond reduction are also explored.

Commentary- student PowerPoint slide-Glutathione- and non-glutathione-based oxidant control in the endoplasmic reticulum. Pathways of thiol-disulfide exchange that involve PDI, Ero1, nascent proteins and glutathione. Thiol groups (carbon-bonded SH-groups) in nascent secretory or membrane proteins (PSH) and glutathione (GSH) enter the ER from the cytosol and react with PDIox, yielding the disulfide-bonded proteins (PSSP), GSSG, and PDIred. Correctly folded PSSP and GSSG are exported to the Golgi complex by vesicular transport. PSSP with non-native disulfide pairings (which are, therefore, misfolded) can be reduced by PDIred (or by other reduced PDI family members or by GSH directly; not depicted), yielding PSH and PDIox. Alternatively, PDIredis recycled to PDIox by reacting with the active form of Ero1α that passes the electrons on to molecular oxygen (O2) [referred to as Ero1α-driven oxidation of PDIred or de novo disulfide (S–S) generation; green arrow]. Ero1α is positively regulated by a high concentration of PDIred(yellow arrow). H2O2, which is generated by Ero1α for every PDIredmolecule that is oxidized, is also converted to water by oxidizing PDIred(see text for details). When PDIox, GSSG and PSSP become abundant, the high PDIox: PDIred ratio promotes the formation of regulatory disulfides in Ero1α (light-blue arrow) that render Ero1α inactive. In addition, a high concentration of GSSG facilitates GSSG-driven oxidation of PDIred(dark-blue arrow). In some tissues, Ero1α is complemented by Ero1β, which is also redox regulated (Wang et al., 2011). http://jcs.biologists.org/
Pathways of thiol-disulfide exchange that involve PDI, Ero1, Nascent proteins and Glutathione:
Thiol groups (carbon-bonded SH-groups) in nascent secretory or membrane proteins (PSH) and glutathione (GSH) enter the ER from the cytosol and react with PDIox, yielding the disulfide-bonded proteins (PSSP), GSSG, and PDIred. Correctly folded PSSP and GSSG are exported to the Golgi complex by vesicular transport. PSSP with non-native disulfide pairings (which are, therefore, misfolded) can be reduced by PDIred (or by other reduced PDI family members or by GSH directly; not depicted), yielding PSH and PDIox. Alternatively, PDIred is recycled to PDIox by reacting with the active form of Ero1α that passes the electrons on to molecular oxygen (O2) [referred to as Ero1α-driven oxidation of PDIred or de novo disulfide (S–S) generation; green arrow]. Ero1α is positively regulated by a high concentration of PDIred (yellow arrow). H2O2, which is generated by Ero1α for every PDIred molecule that is oxidized, is also converted to water by oxidizing PDIred (see text for details). When PDIox, GSSG and PSSP become abundant, the high PDIox: PDIred ratio promotes the formation of regulatory disulfides in Ero1α (light-blue arrow) that render Ero1α inactive. In addition, a high concentration of GSSG facilitates GSSG-driven oxidation of PDIred (dark-blue arrow). In some tissues, Ero1α is complemented by Ero1β, which is also redox regulated (Wang et al.).
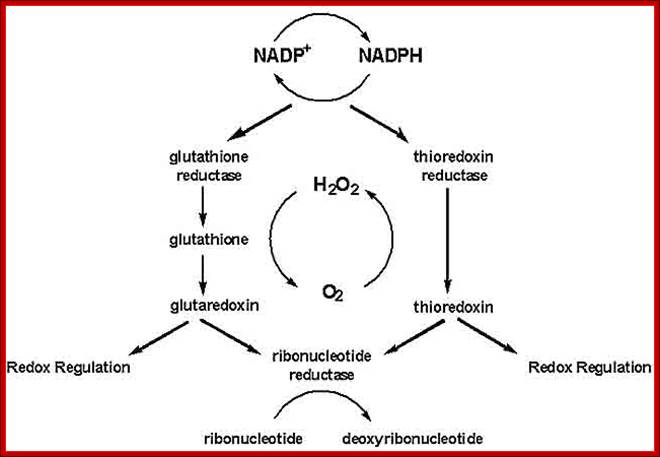
Pathways for disulfide bond reduction in the cytoplasm of E.coli. To disrupt both pathways the most common route is to knock-out the gor and trxB genes. There are already over 80 human proteins directly implicated in protein folding in the ER; http://www.bioscience.org/
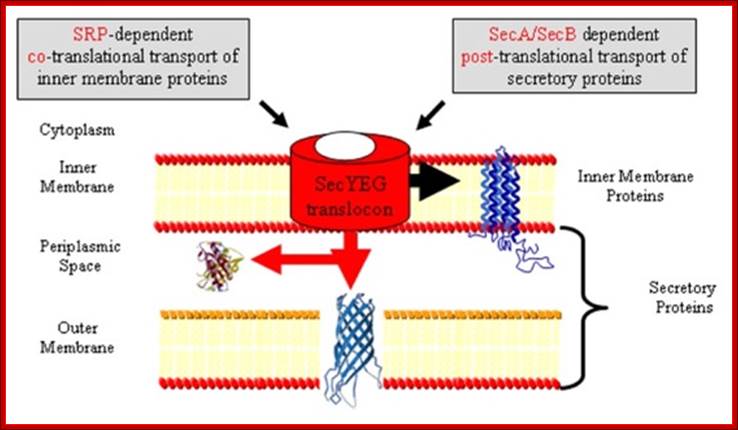
Protein Transport / Secretion in Bacteria:
In gram-negative bacteria, secretory proteins have to be transported across the inner and outer membranes. Bacterial systems have employed both co-translation and post translation methods in transporting proteins.
- Proteins that are destined to be exported contain a hydrophilic region at N-terminal region and a hydrophobic core adjacent to it.
- Several gene products control secretary functions, and they are called Sec proteins.
- While transporting, proteins are unfolded and held in the same pattern in transit; once they are transported into periplasm; proteins assume their normal conformation. E.g. When B-lactamase, while it is transported it is sensitive to Trypsin, but once it is transported out, it becomes Trypsin resistant for it is folded.
E. coli Signal Sequences from the Swiss-Prot Database:
|
Entry |
Description |
Sequence |
|
P31550 |
THIAMIN-BINDING PERIPLASMIC PROTEIN PRECURSOR |
MSAPAVAVTAPVFA |
|
P29679 |
ACYL-COA THIOESTERASE I PRECURSOR (PROTEASE I) |
MMNFNNVFRWHLPFLFLVLLTFRAAA |
|
P19935 |
TOLB PROTEIN PRECURSOR |
MKQALRVAFGFLILWASVLHA |
|
P10904 |
GLYCEROL-3-PHOSPHATE-BINDING PERIPLASMIC PROTEIN PRECURSOR |
MKPLHYTASALALGLALMGNAQA |
|
P37387 |
D-XYLOSE-BINDING PERIPLASMIC PROTEIN PRECURSOR |
MKIKNILLTLCTSLLLTNVAAHA |
Bacterial signal sequences (some):
· Coat-protein: lys.lys.ser.val.leu.lys.ala.ser.val.ala.val.ala.thr.leu.val.pro.met.leu.ser.phe.alla.I ala.gln.
· Periplasmic protein:
· MKQSTIALALLPLLPTPVTKA I RT.
· Beta lactamase:
· MSIQHFRVALIPFFAAFCLPVFA I HP.
· Lipoprotein:
· MKATKLVLGAVILGSTLLAG I CS.
· Archaeal signal sequences:
· MTKLKDQTRAILLATILMVTSVFAGAIAFTGSAAA,
· MLELLPTAVEGVS,
· MVASALATGVFA
Sec YEG protein complex in Transport across Bacterial inner membrane:
- Several Sec proteins are found anchored in the inner membrane and they are involved in transport of proteins.
- Sec Y, sec-E and Sec-G form a channel called Translocon, located in the membrane. They are assisted by Sec C and Sec D
- Sec-A, another ATPase protein, is located at the inner surface of the translocon in the membrane but bound to sec-Y. Yet another protein sec-B recycles between the membrane and in free-state; acts like a chaperone.
- Sec-Y is a transmembrane with ten membrane passes and it has ATPase activity.
- The sec-Y is like Sec 61 of eukaryotic system, where it acts like a translocon.
- Towards the periplasmic side of the translocon; there are three more subunits such as D and F and YajC periplasmic but anchored to membrane.
- On the left of SecY group, another subunit is associated called Yid C acts as insertase, works with Sec translocon.
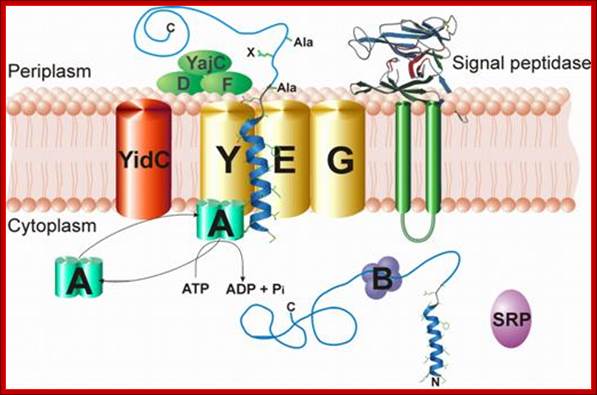
Protein transporters in bacterial system, e.g. E.coli, transporters are located in its cell membrane and protein is transported into periplasmic space. SeC- B dependent pathway. From SRP proteins B picks up by the NH2 terminal part of the protein and then it hands over to Sec YEG; http://neutrons.ornl.gov/; http://www.sfu.ca/mbb/mbb/faculty/pa; etzel/Mark_Paetzel_Page_files/IMAGE002.JPG
Sec dependent protein secretion; The function of the bacterial signal recognition particle (SRP) receptor in co-translational targeting to the SecYEG translocon: Co-translational targeting of a bacterial membrane protein requires the SRP-dependent recognition of a membrane protein when it emerges from the ribosomal exit tunnel and the subsequent targeting to the SecYEG translocon. Here, the SRP receptor plays a crucial role because it provides the essential link between the soluble SRP-ribosome-nascent chain complex (SRP-RNC) and the membrane bound SecYEG translocon. We are interested in understanding how the bacterial SRP receptor binds to the membrane and how membrane binding is coordinated with the SRP-dependent targeting, Prof. Dr. Hans-Georg Koch. http://www.biologie.tu-dresden.de/
- At the outer surface, i.e. in periplasmic space, the translocon is bound to signal peptidase.
- Proteins required for plasma membranes and several secretary proteins are all synthesized on free ribosomes and none of them are bound to any membranes.
- Sec-B acts as a chaperone protein that helps newly synthesized proteins to move towards the channel region. First sec-B associates with the nascently synthesized protein, where the proteins are folded in a different mode. In this complex, sec-B takes the protein to the inner region of the channel proteins, where it interacts with sec-A. Sec-A using ATP energy drives the protein through the channel with signal sequence forward. Sec-D and Sec-F provide motor function and assist Sec-A.
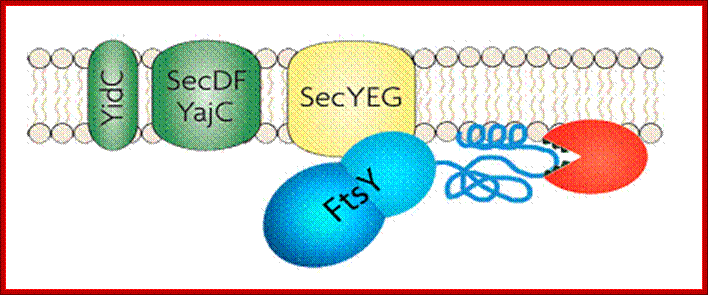
Membrane binding of FtsY, the bacterial SRP-receptor. Binding is mediated by protein-protein interaction via the SecYEG complex and by protein-lipid interaction. (for details see Weiche et al., 2008), Prof. Dr. Hans-Georg Koch.
- Proteins that are transported have N-terminal hydrophobic signal sequences and adjacent hydrophilic core sequences.
- As the signal sequence passes through, the translocon, the signal sequences are cleaved by signal peptidase and the protein is drawn into periplasmic space. An electron motive force that develops during translocation helps in protein transport across the membrane.
- As the protein is threaded though, sec-B is released, then SecB picks another protein to transport.
- Several chaperones increase the efficiency of secretary transport system, e.g. GroEL, and GroEC.
SRP mediated Co-translational Export of Proteins:
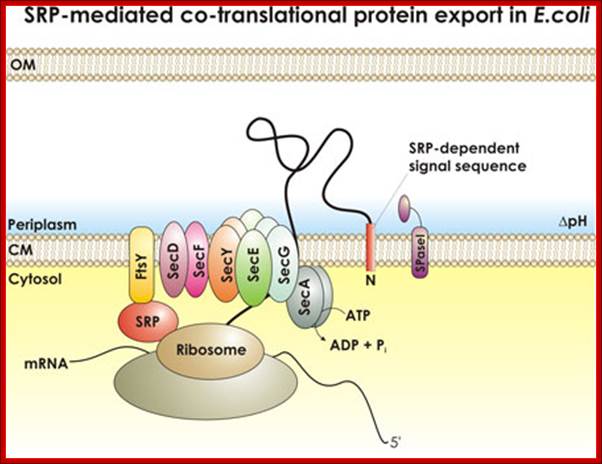
SRP mediated transport; http://beckwith.med.harvard.edu/
The SecYEG–SecDFYajC complex is the general translocase that is used by Escherichia coli to insert newly synthesized polypeptides into the cytoplasmic membrane. The signal-recognition particle (SRP) and SRP receptor (FtsY) target most Sec-dependent proteins. The novel insertase YidC can function with the Sec translocase or independently to promote membrane protein insertion. Some Sec-independent proteins are also targeted by the SRP. E. coli 4.5S RNA and P48 have been shown to be homologous to eukaryotic SRP7S RNA and SRP54, respectively.

Protein translocation through the SecYEG complex. (A) Schematic overview of protein translocation and membrane protein insertion through the SecY complex in bacteria. Nascent chains having a signal sequence (red) emerge from the ribosomes (grey) and are localized to the SecY complex (green) co-translationally by the signal recognition particle (SRP) and FtsY/SRP receptor (SR; left). Alternatively, they can be presented post-translationally through SecA or SecB (right). (B) Schematic of two possible dimeric conformations of SecYEG, shown looking down onto the membrane. In the back-to-back form, the SecE transmembrane domains (orange cylinders) are juxtaposed and the lateral gates face outwards; the front-to-front configuration places the lateral gates close to one another. (C) Ribbon representation of the Methanococcus jannaschii SecYEβ structure, solved by X-ray crystallography (Protein Data Base file 1RHZ; van den Berg et al, 2004): the side view is from within the plane of the membrane and the top view is from the cytoplasmic side. SecY is shown in green, with the two halves of the clam shell in light and dark shades. SecE and Secβ (SecG) are shown in red and blue, respectively. The plug helix of SecY is shown in orange and an arrow indicates the lateral gate. (D) Homology model of the dimeric, membrane-bound Escherichia coli SecYEG structure, built using coordinates of the M. jannaschii structure and then docked into the electron microscopy map from the membrane-bound two-dimensional crystals (only the membrane-bound domains were modelled; Bostina et al, 2005). SecY (green), E (red/orange) and G (blue) are shown in different shades for the two monomers. Residue Leu 106 in SecE, at which cysteine crosslinking between monomers can be achieved, is shown as spheres. The membrane is shown in grey, Alice Robson & Ian Collinson.
- The SRP pathway consists of eight protein subunits secD, secF, secY, secE, secG and Sec A,. Srp receptor is a transmembrane protein anchored. Sec A located at the inner surface of the Sec DFYEG complex. Though the structure is complex, this pathway is extensively used in protein translocation. SecY, Sec E and Sec G form translocon.
In Escherichia coli 4.5 S RNA is metabolically stable and abundant RNA. It consists of 114 nucleotides, and it is structurally homologous to domain IV of mammalian signal recognition particle (SRP) RNA (7sLRNA). It is now known that one of the 4.5 S RNA-binding proteins (a gel mobility shift assay) has been identified as Ffh, which has been characterized. It is similar to 7sSRP 54 subunit.
- The Fts-Y protein (45kDa?) (like α subunit of SRP receptor protein), is an integral membrane protein acts as a receptor. They recognize anchor sequences and transport proteins into membranes. Another 48 kd protein found there also helps in co-translational secretary process. SRP mediated process is an efficient translocation system.
The signal recognition particle (SRP) is a universally conserved RNA-protein (ribonucleoproteins) complex involved in the transport and targeting of proteins to the cell membranes. The RNA and protein components of this complex are highly conserved but do vary between the different kingdoms of life.
The eukaryotic SRP consists of a ~300-nucleotide long RNA (known as 7SL) and six proteins known as SRPs 72, 68, 54, 19, 14, and 9. Archaeal SRP consists of a 7S RNA and homologues of the eukaryotic SRP19 and SRP54 proteins. In most bacteria, the SRP consists of an RNA molecule (4.5S) and the Ffh protein (protein fifty four homologue) a homologue of the eukaryotic SRP54 protein. Some Gram-positive bacteria (e.g. Bacillus subtilis) have a longer eukaryote-like SRP RNA that includes an Alu domain. The Signal Recognition Particle Database (SRPDB) provides compilations of SRP components.
![]()
Fig. Schematic overview of the bacterial protein translocation system termed Sec translocase. Proteins which are synthesized within the ribosome are exported from the cytoplasm over the cytoplasmic membrane into the periplasm by the proton motive force (PMF) and the ATPase SecA via the heterotrimeric membrane channel SecYEC. Two major ways of protein translocation exist: posttranslational secretion and cotranslational insertion into the cytoplasmic membrane. Secretory proteins are either directly targeted to SecA by means of their N-terminal signal sequence or are bound by the chaperone SecBfirst and translocate later via SecA and SecYEG. Membrane proteins are translocated cotranslational. Their C-terminal signal sequence is bound by the signal recognition particle (SRP) and targeted to the SRP receptor (FtsY). Consequently SecAtranslocates these proteins via a lateral gate in the SecYEG channel into the membrane. SecDF(yajC) is an accessory factor which seems to improve preprotein translocation. YidC associates with the translocon during protein insertion into the membrane (figure derived from (1)). Prof. A.J.M. Driessen (Molecular Microbiology
Bacterial Secretary System-Three pathways:
In recent years new dimension has emerged in re of protein secretion in E. coli and other Gram-negative bacteria. The process of protein secretion is complex. In Gram-negative bacteria secreted proteins must cross two membranes employing one of the five different translocation mechanisms. Each mechanism requires the involvement of several proteins.

Diagram of the three Types of secretion pathways of E. coli: In the left of the above diagram one can see SEC mediated pathway, the middle one shows SRP mediated pathway and the rightmost shows Twin Arginine Translocation (TAT) pathway which contains TAT translocon. http://www.Image.3air.net
With regard to secretory systems there are at least four types. The type I system is a single-step process where translocation bypasses the periplasmic space. The process requires five proteins, HlyA, HlyB, HlyD, an ABC transporter and TolC. The HlyB/D are inner membrane proteins bound to the ABC transporter. This transporter spans the two membranes and binds two TolC molecules on the outer membrane. The C-terminal portion of HlyA is sufficient to effect translocation through the HlyB/D-ABC-TolC complex and, when fused to a target protein, will direct secretion of the protein to this system.
TAT pathway (Twin Arginine Transport):
Protein translocation through the inner membrane is accomplished by SecB-dependent (SEC) pathway, signal recognition particle (SRP) pathway and the third TAT pathway. Most proteins in E. coli are exported to the periplasm via SEC system. Extracellular secretion by a type II mechanism is via the main terminal branch pathway, which involves a poorly defined complex of 12-16 proteins that are not expressed under normal laboratory conditions. Therefore, most of the proteins exported by this pathway remain resident in the periplasm, which is also where most secreted recombinant proteins accumulate. The SRP pathway is cotranslational and Sec and TAT are post translational pathways.
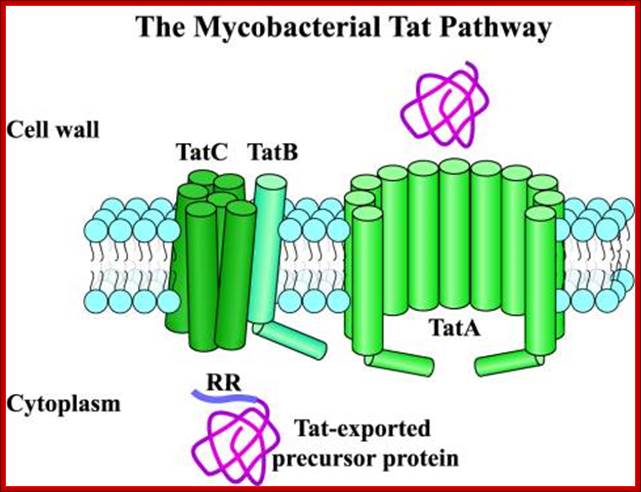
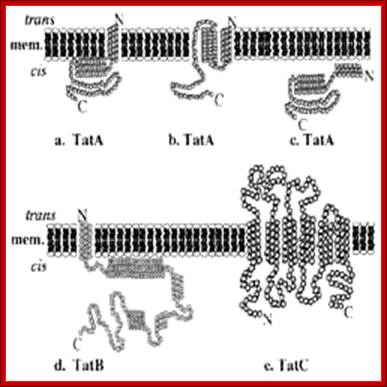
The Twin-Arginine Transport System (TAT); http://www.landesbioscience.com/
The twin-arginine transport (TAT) system is a protein-targeting pathway found in the cytoplasmic membranes of many eubacteria, some archaea, and the chloroplasts and mitochondria of plants. It is apparently not a feature of animal physiology. Substrate proteins are targeted to a membrane-bound transport apparatus by N-terminal signal peptides harboring a distinctive ‘twin-arginine’ amino acid sequence motif, and most remarkably, all substrate proteins are transported in a fully folded conformation. Model systems most commonly used to study the fundamentals of TAT transport are the Gram-negative eubacterium Escherichia coli, the Gram-positive eubacterium Bacillus subtilis, and thylakoid membranes derived from pea or maize chloroplasts. Here, we have attempted to integrate our knowledge of the key aspects of these well-characterized Tat protein transport pathways, to carve-out some shared principles between systems, and arrive at a broad consensus covering the physiology and biochemistry of Tat transport: Frank Sargent, Ben C. Berks and Tracy Palmer.
|
Group and characteristics |
Name |
Size(kDa) |
Location |
Presence in other bacteria |
|
Secretory Chaperonin |
SecB |
18 |
cytoplasm |
wide spread in members of Enterobacteriaceae |
|
General Chaperonins |
DnaK |
69 |
cytoplasm |
universal |
|
Secretory ATPase |
SecA |
102 |
cytoplasm, ribosome, peripheral cytoplasmic membrane |
B. subtilis, wide spread in members of Enterobacteriaceae |
|
Translocase |
SecD |
67 |
cytoplasmic membrane |
not identified |
|
Signal Peptidase |
LepB |
36 |
cytoplasmic membrane |
S. typhimurium, |
|
Others |
Ffh |
48 |
cytoplasm |
not identified |
A list of proteins involved in bacterial transport pathway:
Transport of Bacterial and Eukaryotic proteins:
The Sec translocase can participate in co-translational translocation during which the nascent protein is translocated while the ribosome is bound to the central core of the Sec translocase, the protein conducting channel (PCC). The Sec translocase also participates in post-translational translocation, in which a translated unfolded protein is pushed or pulled through the PCC by energy-using soluble components, such as SecA in bacteria (a) and BiP in eukaryotes (d).
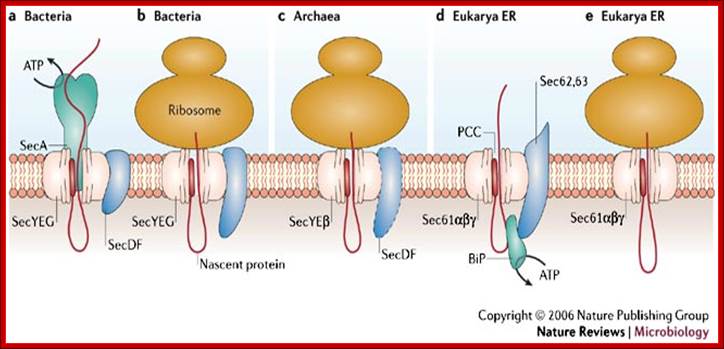
Protein secretion in the Archaea: multiple paths towards a unique cell surface; The Sec translocase can participate in co-translational translocation during which the nascent protein is translocated while the ribosome is bound to the central core of the Sec translocase, the protein conducting channel (PCC). The Sec translocase also participates in post-translational translocation, in which a translated unfolded protein is pushed or pulled through the PCC by energy-using soluble components, such as SecA in bacteria (a) and BiP in eukaryotes (d). The main components of the Sec translocase are shown for bacterial post-translational translocation (a) and co-translational translocation (b). Co-translational translocation in archaea is shown in (c). Post-translational translocation and co-translational translocation in the endoplasmic reticulum (ER) of the eukaryote Saccharomyces cerevisiae are shown in (d) and (e), respectively. The Sec translocase in archaea has mosaic features of the Sec translocase in both bacteria and the endoplasmic reticulum; http://www.nature.com/
The main components of the Sec translocase are shown for bacterial post-translational translocation (a) and co-translational translocation (b). Co-translational translocation in archaea is shown in (c). Post-translational translocation and co-translational translocation in the endoplasmic reticulum (ER) of the eukaryote Saccharomyces cerevisiae are shown in (d) and (e), respectively. The Sec translocase in archaea has mosaic features of the Sec translocase in both bacteria and the endoplasmic reticulum.
DNA transport during sporulational cell partition:
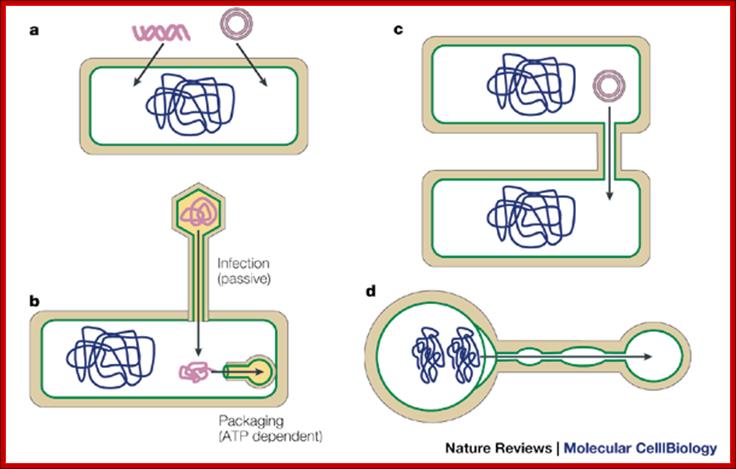
Bacterial viruses (bacteriophages) also need to transport their genomes into cells. Although a range of strategies are used for DNA transport during infection, the rate of transport is generally much higher than for transformation or other mechanisms, and it probably occurs through facilitated diffusion. During infection, phage DNA is translocated through a channel or pore consisting of both phage and host proteins, or only phage proteins49. At the end of the infectious cycle, another DNA transfer process is needed, to package the newly replicated phage genomes into the preformed procapsids. This process requires energy, and it is unique in that the barrier crossed by the DNA molecule is not a membrane but the pore of a transport protein called the 'portal protein' or 'head–tail connector; Jeff Errington, Jonathan Bath & Ling Juan Wu http://www.nature.com/
During sporulation in B.subtilis, cell division starts with septal formation unequally and produces small forespore cell and large mother cell. Even before this event the DNA has already replicated. During the transportation of one of the two daughter DNA molecules across the septum, SpoE III plays an important role. The factor associated with other protein factors recognizes SpoIIIE receptor region on the DNA and then transfers one of the daughter DNA with Ori C in fore front.
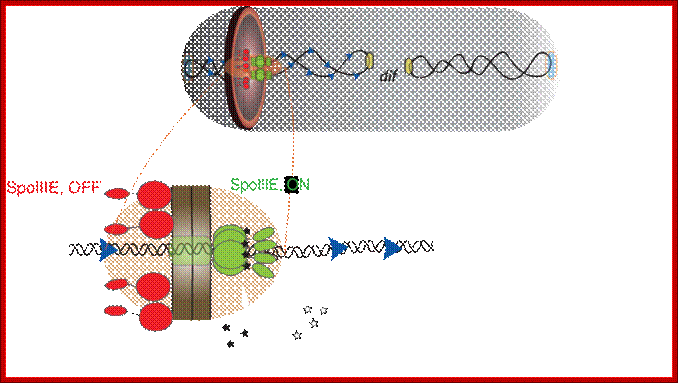
Schematic of a sporulating B. subtilis cell (gray) at the
onset of DNA translocation, in which each chromosomal arm (black ribbon) is
bound by two opposing active unidirectional complexes (green) that assemble on
each side of the division septum (brown disc). Below, an enlarged view of a
single chromosomal arm (helix), division septum (gray) and SRS sequences (blue
triangles) are represented. Black arrows depict the overall direction of DNA
transport. We assume that during translocation DNA is transported in the
direction from SpoIIIE-![]() to SpoIIIE-
to SpoIIIE-![]() ; SpoIIIE-
; SpoIIIE-![]() –mediated interactions with nonpermissive SRS in the forespore leads
to the inactivation of the forespore complex (red). The inactivation of the
forespore SpoIIIE complex converts the bidirectional channel into a DNA
exporter, leading to unidirectional DNA transport into the forespore.
The transport complex holds ori c region and pulls the bacterial DNA across the
fore spore septum. Jerod L Ptacin1, Marcelo Nollmann1,, Eric C Becker, Nicholas R
Cozzarelli1, , Kit Pogliano & Carlos Bustamante.
–mediated interactions with nonpermissive SRS in the forespore leads
to the inactivation of the forespore complex (red). The inactivation of the
forespore SpoIIIE complex converts the bidirectional channel into a DNA
exporter, leading to unidirectional DNA transport into the forespore.
The transport complex holds ori c region and pulls the bacterial DNA across the
fore spore septum. Jerod L Ptacin1, Marcelo Nollmann1,, Eric C Becker, Nicholas R
Cozzarelli1, , Kit Pogliano & Carlos Bustamante.
DNA transfer in extreme thermophilic bacteria:
Thermophilic bacteria were found to clearly stand out in terms of inter-domain DNA transfer, such as 24 and 16.2% of the genes in the hyper-thermophilic bacteria Thermotoga maritima and Aquifex aeolicus, respectively, are suggested to be transferred from archaeal hyper-thermophiles. Despite the massive inter-domain gene transfer between hyper thermophilic archaea and bacteria until recently nothing was known with respect to the components of the transformation machineries of extremely thermophilic bacteria. To get insights into the transformation machinery of extremely thermophilic bacteria we chose the transformable strain Thermus thermophilus HB27, which was subject of whole genome sequence analysis by the Göttingen genomics laboratory. Authors got access to the sequence data and performed a genome based approach to search for genes involved in DNA uptake in Thermus. This approach led to the identification of the first components of the DNA transformation machinery in a thermophilic bacterium.
Currently focus is on functional analyses of 16 identified proteins of the DNA translocator, and the structure, function, and regulation of the DNA translocator in Thermus. These studies shed the first light onto the structure and function of the Thermus transformation machinery and led to the first model of a DNA translocator in thermophilic bacteria.
Further studies are underway to address the structure of the DNA translocator. Furthermore we will analyze the horizontal gene transfer between different domains, such as extremophilic archaea and bacteria, and unravel the impact of transformation systems of extremophilic bacteria in the evolution of prokaryotes.
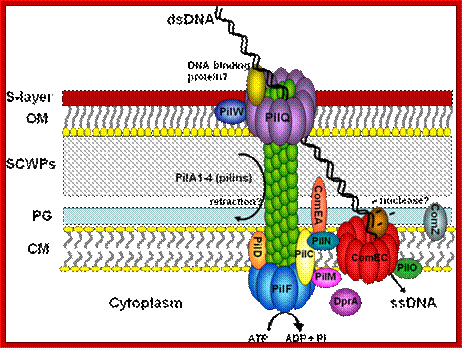
DNA transfer in mesophilic bacteria; ;http://www.uni-frankfurt.de/
Type III –TTSS pathway:
|
|
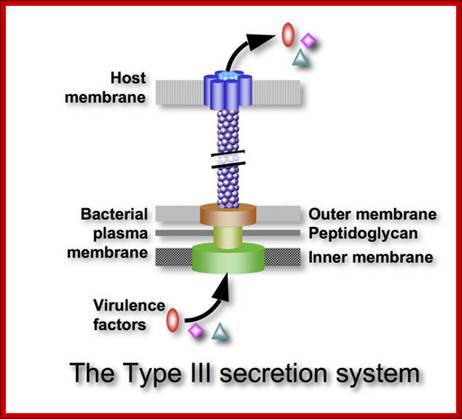
{kind=link}
Combined X-ray crystallography and electron cryo-microscopy was used to investigate structures of large molecular assemblies at atomic resolution. http://www.educatetruth.com/
Many Gram-negative pathogenic bacteria employ a complex protein secretion system termed type III secretion system (TTSS) to transport bacterial effector proteins across three membrane barriers into eukaryotic host cytoplasm. The effector proteins delivered by TTSS are capable of modulating and interfering with the host cellular processes, which cause diseases in animals and plants such as plague, typhoid fever, bacterial dysentery. The TTSS is composed of more than 20 structural proteins, effector proteins, and chaperones. Structural investigation targets the major structural components, the needle complex and the translocon, of TTSS in Shigella flexneri. We combine X-ray crystallography and electron cryo-microscopy to investigate structures of large molecular assemblies at intermediate to atomic resolution. Agrobacterium tumefaciens transports its tumor causing DNA and induce cell transformation into tumors in plants. For plant genetic engineering transformation Agrobacterial plasmid is recombined for the convenience of plant molecular biologists (authors ??).
Few Rhizobium (Agrobacterium) bacterial strains transfer
Genes into plant cells:
The strains like LBA 4404 have plasmids, which lack tumor inducing genes, but they do contain a restrained Ti plasmid with Vir genes, which can provide components for transferring DNA segments that are found in other plasmid DNA, if located in between the LB and RB. Genes such as Shi, Roi and Opine producing genes are deleted from these plasmids. The said strains also lack in endonuclease activity (Rec ^-- RE ^ - ) and they are sensitive to some antibiotics such as Carboxylin.
Kingdom: Bacteria,
Phylum: Proteobacteria,
Class:Alpha Proteobacteria,
Order; Rhobiales;
Family:Rhobiaceae
Genus ;Rhizobium,
Species: radiobacter
(old name: Agrobacterium tumefaciens),
11 August 2011
Agrobacterium rhizogenes:
Phylum: Proteobacteria,
Class-Alphaproteobacteria,
Order: Rhizobiales,
Genus: Agrobacterium,
Species: rhizogenes.
The above bacterial plasmid when introduced into plant cells, causes adventitious multi-branched hairy root initiation in large numbers (in Dicots).
Agrobacterium’ Ti plasmid:
Ti plasmids range in size from ~200 to ~225 kbp. Of this, about 30% is common to Nopaline and Octopine plasmids (Genes VIII, Benjamin Lewin, pp. 525). The Ti plasmids are classified into different types based on the type of opine produced by their genes. The different opines specified by pTi are Octopine, Nopaline, Succinamopine and Leucinopine (Wikipedia). The plasmid has 196 genes that code for 195 proteins. There is no structural RNA genes in the plasmid. The plasmid is ~2,06,479 nucleotides long, the GC content is 56% and 81% of the DNA has coding genes. There are no pseudo genes.
Types of Ti Plasmids
Two classes of Ti-plasmids exist. The Nopaline and Octopine Ti-plasmids have T- DNA regions but the structure of these regions differs.
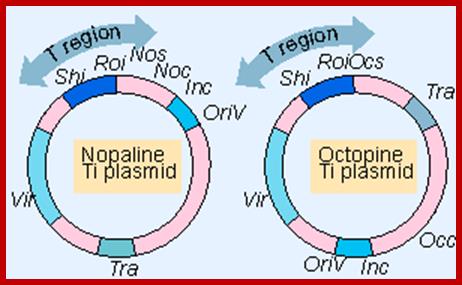
Nopalkine and Octopine Ti plasmids carry variety of genes including regions that have overlapping functions;

http://argo-mlm.ru
The TDNA of Ti plasmid transferred into plant cells induce the synthesis of certain low molecular weight compounds which are released from pant cells and they are used and catabolised by the bacteria.
A part of DNA that is transferred from Ti plasmid DNA of bacteria into plant cell is called T-DNA is referred to as the T-region. T-region is approximately 10 to 30 kbp in size and represents less than 10% of the Ti plasmid. Ti plasmids generally contain one T-region, but some Ti plasmids have multiple T-regions in them. T-region is flanked by T-DNA left and right border repeats. These are ~25 bp long and highly homologous in sequence. The T-DNA border sequences flank the T-region in directly repeated orientation. The T-DNA border sequences delimit the T-DNA and are the target of the border-specific endonucleases. The sequences are
TG Pu G ATTGGCAGGATATAT N TGTAANC C T TC
The Ti plasmid consists of a replication origin OriT. It has Opine catabolytic gene. It also contains Virulent genes (Vir), Bacterial chromosomes contain important regulatory genes such as Chv.
A part of TDNA – from the left->
Vir = Virulence regulators left of LB.
LB = Left border.
iaaH = (aux2 and tms 2), Indole-acetamide hydrolase gene which converts IAM to IAA. iaaM = (aux 1, tms 1, Tryptophan-2mono-oxygenase, which converts Tryptophan into indole3-acetamide (IAM).
IPT = Isopentinyl transferase gene.
Nos = Nopaline synthase gene.
RB = Right border.
o/D = Over drive sequence right of RB.
The said plasmid genes are involved in the synthesis of Auxin and Cytokinin.
The Ti plasmid, outside the T-DNA region, also contains several other genes such as opine catabolizing genes such as Nopaline catabolizing gene (NOC), Octopine catabolizing gene (OCC) and Agropine catabolizing gene (AgC). On to the left of the T-DNA there is a segment of DNA, 36kbp, consisting of a cluster of genes called ViR (virulence) genes. On the right flank of the RB of T-DNA abutting to it is a 25 bp long sequences called Over- Drive (5’TAApuTpyNCTGTpuTNTGTTTTGTTTG3’). This region is recognized by ViR-C gene products and facilitates ViR-D2 gene products to cut the strands in the RB.
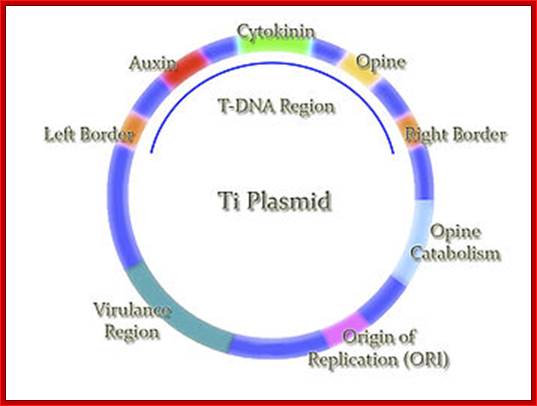
A general view showing T DNA and regions such as opine genes, Ti origin for replication and Vir genes. http://en.wikipedia.org/
The receptor that responds to signal is VirA. It has autokinase activity and phosphorylates at serine. Then the activated VirA-P phosphorylates VirG found at the cytosolic side of the VirA. Both genes, on Ti plasmid, are constitutively expressed. The activated G is a transcriptional factor (TF). It activates the rest of the Vir genes.
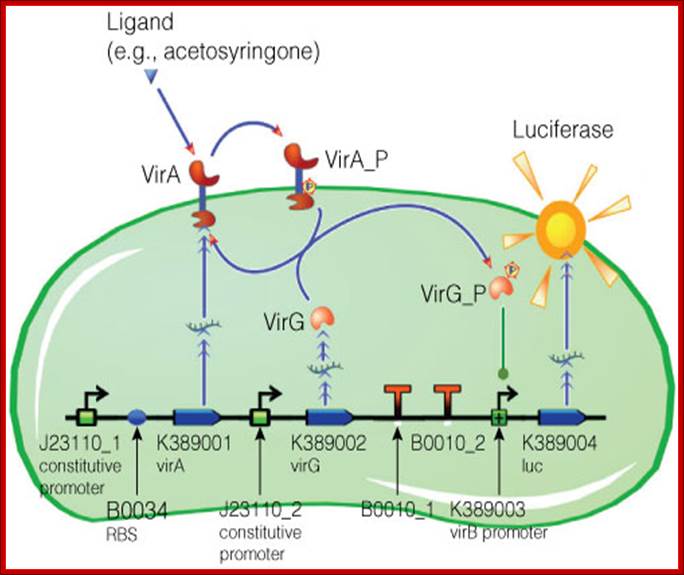
Figure: Model of acetosyringone inducible luciferase expression in E. coli. VirA and VirG are constitutively expressed. Once activated by VirA, it acts as transcriptional activator of other Vir genes. http://www.promega.es/
Vir Genes and their Functions: |
|
Vir Gene |
Function |
|
Vir A, |
A receptor with two component system reacts from wounded plant cells and phenolic compounds such as acetosyringone, syringealdehyde or acetovanillone which leak out of damaged plant tissues. VirA gets activated with the bindng ofd signals and autophosphorylates and inturn phosphorylates VirG
|
|
VirG |
VirG is Transcriptial factor, p-lated VirG induce expression of other virulence genes. |
|
VirD2 |
Endonuclease; cuts T-DNA at right border to initiate T-strand synthesis |
|
Vir D1 |
Topiosomerase; Helps Vir D2 to
recognise and cleave within the 25bp |
|
Vir D2 |
An endonuclease nicks the 5’end
of the T-DNA at the RB and covalently attaches to the 5’ end of the T-strand,
thus forming the |
|
VirD4 |
VirD4 is the ATP-dependent linkage of protein complex necessary for T-DNA translocation
|
|
Vir C1 |
Binds to the 'overdrive' region
to promote high efficiency T-strand
|
|
Vir E2 |
Binds to T-strand protecting it
from nuclease attack, and intercalates |
|
Vir E1 |
Acts as a chaperone which stabilizes Vir E2 in the Agrobacterium. It is essential for the export of E2 into the plant cell and into the nucleus |
|
Vir B & Vir D4 |
Assemble into a secretion system which spans the inner and outer bacterial membranes. Required for Export of the T-complex and Vir E2 into the plant cell |
|
VirBs |
Proposed- VirB7, VirB8, VirB9, and VirB10 are the primary constituents of the T-DNA transport pore. VirB7, a lipoprotein, is anchored to the outer membrane (13), while VirB8 and VirB10 are inner membrane proteins. , and VirB10 participate in the formation of oligomeric complexes. These studies support the proposed role of the VirB7 to VirB10 proteins in transporter assembly. VirB4 and VirB11 are homo- and heterodimers (Dang and Christie, 1997). The VirB7-VirB9 heterodimer is assumed to stabilizes other Vir proteins during assembly of functional transmembrane channel
|
|
Host interacting factors |
AtKAPα, CypA, PP2C, RocA, Roc4 ASK (Skp1-like), VIP1
|
Agrobacterium virulence system:
The T-DNA transfer is mediated by products encoded by the 30-40 kb vir region of the Ti plasmid. This region is composed by at least six essential operons (vir A, vir B, vir C, vir D, vir E, virG ) and two non-essential (virF, virH). The number of genes per operon differs, virA, virG and virF have only one gene; virE, virC, virH have two genes, while virD and virB have four and eleven genes respectively.. VirA and Vir G are constitutively expressed. Others are expressed by the activity of VirG.
Nopaline T-DNA
- single continuous segment of about 22kb
Octopine T-DNA - three segments; left DNA
(TL) = 13 kb; center DNA (TC),
ViR-B: It consists of 11 genes called ViR-B1 to ViR-11. They are located in plasma membrane. They perhaps organize into channel like structure and involved in the transport of T-DNA in single strand form. The B-11 has ATPase activity.

https://web.williams.edu
VirB protein complex (11 proteins) act as transporter of T DNA from bacteria to plant cells; This process is deemed as T4ss type; Recent work from several labs, including my own, supports the notion that assembly of a membrane-associated heteroligomeric VirB protein complex is a multistep process that is sensitive both to environmental influences and to the presence of nonfunctional constituent subunits. Over the past few years, we have focused on the role of several VirB proteins, and particularly VirB10, in transport apparatus assembly. Although various lines of experimentation implicate VirB10 as an essential and perhaps rate-limiting component of the transfer process, the precise activity of the protein is unknown. We have used both genetic and biochemical strategies to characterize VirB-containing protein complexes and the interactions that mediate VirB protein function. Through our biochemical studies, we have found that VirB10 stability is dependent on growth temperature and periplasmic osmodaption. Our genetic experiments have led us to conclude that over-production of wild-type VirB10 inhibits DNA transfer and suggest a mechanism by which VirB10 accumulation is regulatedin vivo. Additionally, we have isolated and characterized several avirulent virB10point mutants. https://web.williams.edu
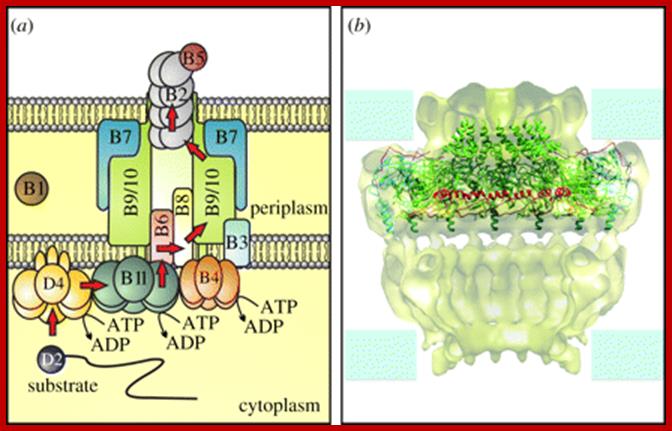
General translocation pathway and core structure of a T4SS. (a) The translocation pathway of substrates through the Agrobacterium VirB/D4 system. For a detailed description see text. (b) A cutout of the Type IV secretion system core complex and crystal structure of the O-layer. The cryo-EM structure (surface representation) of the T4S system core complex is here superimposed with the crystal structure of the O-layer. For the crystal structure, VirB7, the C-terminal domain of VirB9 and the C-terminal domain of VirB10 are shown in magenta, cyan and green. The lever arm sequence of VirB10 is shown in red. Figure contributed by Drs Karen Wallden and Gabriel Waksman.http://rstb.royalsocietypublishing.org/
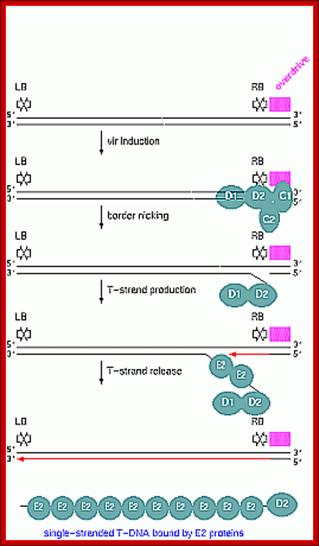
Once the Ti plasmid genes are activated, the VrC1 binds to the RB over drive sequence; this leads to the assembly of VirD2 and endonuclease assemble on to the 25bp long RB and cuts one strand of the DNA at 5’ end releases one strand and the VirD2 covalently bind to the 5’ of ss DNA strand. The strand that is cut and removed is replaced by DNA polymerase that extends the 3’end of the cut end.
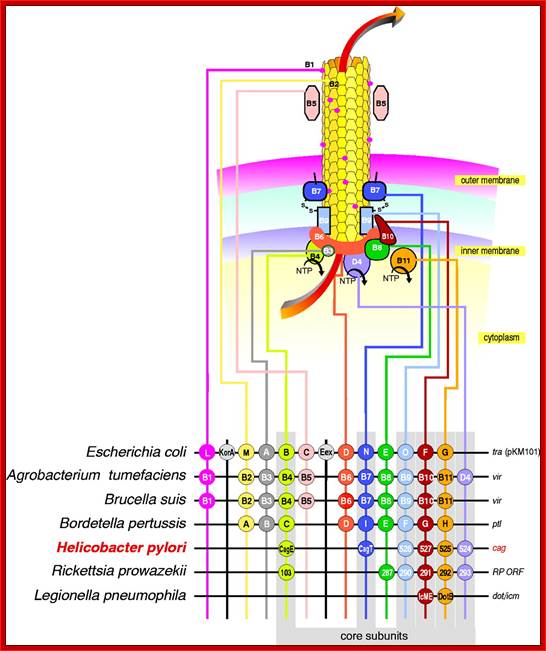
T4SS system, The above mentioned channels are a paradigm for type IV secretion systems (T4SS). http://igem-victoria.pbworks.com/

Hypothetical model for T-DNA transfer from the bacteria into the plant cell. In the bacterial cell, the VirE1 chaperone prevents binding of VirE2 to ssDNA, as has been suggested (10–12), and possibly prevents VirE2-dependent channel formation in the bacterial membrane. VirE2 is transported through the VirB-VirD4 channel and subsequently inserts into the plant plasma membrane, allowing the transport of the ssDNA-VirD2 complex. The way in which the VirE2 molecules enter the cytoplasm is unclear. However, once in the cytoplasm, VirE2 protein molecules coat the complex, permitting its transfer to the nucleus (1–3). For simplicity, the pilus and its involvement in T-DNA transfer are omitted from the scheme. /http://www.pnas.org
Model for assembly of the Agrobacterium T4SS:
A model of how the 12 proteins (VirB1-B11, VirD4) essential for T4SS form a membrane-spanning channel is presented in Figure 1. The T-DNA is transferred as a single stranded intermediate called the T-strand. In addition, 4 proteins, VirD2, VirE2, VirE3, and VirF are exported by the T4SS to plant cells. A model of how the 12 proteins (VirB1-B11, VirD4) essential for T4SS form a membrane-spanning channel is presented in Figure 1. The T-DNA is transferred as a single stranded intermediate called the T-strand. In addition, 4 proteins, VirD2, VirE2, VirE3, and VirF are exported by the T4SS to plant cells. This model was predicted by genetic and bioinformatics approaches. Current efforts aim to define the topology, location and function of the T4SS. We have recently determined that the multiple T4SS localize in a periodic (potentially) helical pattern around the circumference of the bacterial cell.This localization pattern was determined by deconvolution fluorescence microscopy of GFP fusions to T4SS components and substrates. Figure 2 shows several optical sections through the bacterial cell. This localization pattern was confirmed by immuno-fluorescence microscopy of native T4SS proteins. This localization pattern likely facilitates binding of Agrobacterium to susceptible plant cells. Indeed, if one tags the T4SS with GFP, one can see Agrobacterium binding laterally along their lengths to single plant cells; http://pmb.berkeley.edu/
Finding a way to the nucleus; Stanton B Gelvin.

Models describing the possible roles of VirE2 in targeting T-strands to the plant nucleus; T-strands are processed from the Ti-plasmid by the action of the VirD2 endonuclease, which covalently links to the 5′ end of T-strands and pilots them through the Agrobacterium T4SS and into the plant cell. VirE2 protein separately enters the plant cell through the T4SS. In Model 1, VirE2 initially remains in the plant plasma membrane and ‘picks up’ T-strands as they enter the plant cell [47]. In Model 2, VirE2 enters the plant cytoplasm and interacts with T-strands and VIP1 protein, forming the T-complex. Super-T-complexes can form when T-complex proteins further interact with other secreted Agrobacterium virulence effector proteins, and/or with plant-encoded proteins such as VIP1 [17] and importin α [15••]. Nuclear targeting of super-T-complexes may depend upon the phosphorylation status of VIP1 [33••]. In Model 3, VirE2 enters the nuclear membrane, interacts with VirD2 molecules on the T-strands, and helps ‘pull’ T-strands into the nucleus [46••]. VIP1 may subsequently target super-T-complexes to chromatin to facilitate T-DNA integration into the plant genome, Stanton B Gelvin
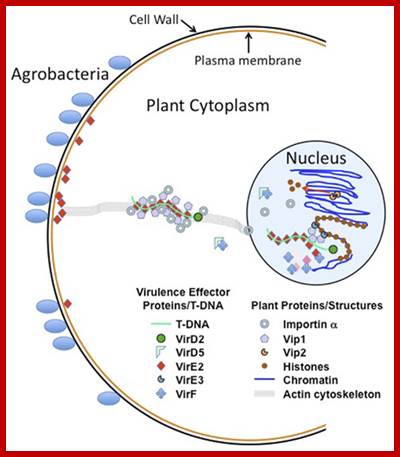
Traversing the cell: Agrobacterium T-DNA’s journey to the host genome; The genus Agrobacterium is unique in its ability to conduct interkingdom genetic exchange. Virulent Agrobacterium strains transfer single-strand forms of T-DNA (T-strands) and several Virulence effector proteins through a bacterial type IV secretion system into plant host cells. T-strands must traverse the plant wall and plasma membrane, traffic through the cytoplasm, enter the nucleus, and ultimately target host chromatin for stable integration. Because any DNA sequence placed between T-DNA “borders” can be transferred to plants and integrated into the plant genome, the transfer and intracellular trafficking processes must be mediated by bacterial and host proteins that form complexes with T-strands. This review summarizes current knowledge of proteins that interact with T-strands in the plant cell, and discusses several models of T-complex (T-strand and associated proteins) trafficking. A detailed understanding of how these macromolecular complexes enter the host cell and traverse the plant cytoplasm will require development of novel technologies to follow molecules from their bacterial site of synthesis into the plant cell, and how these transferred molecules interact with host proteins and sub-cellular structures within the host cytoplasm and nucleus. Stanton B. Gelvin;http://journal.frontiersin.org

https://www.slideshare.net;Plant Transformation methods; https://image.slidesharecdn.com; http://brussels-scientific.com
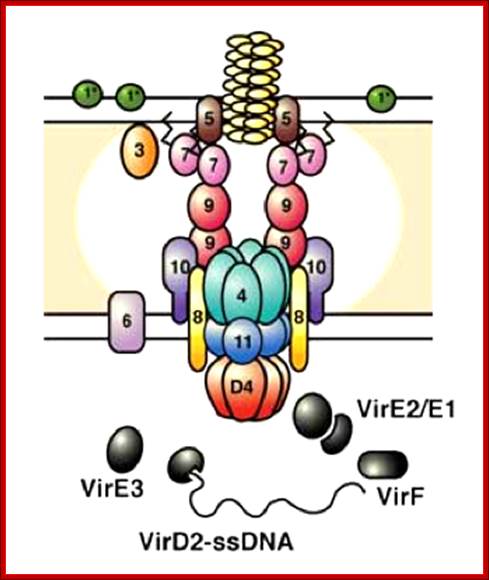
The VirB/D4 Type IV secretion systems (T4SS) of the Agrobacterium as a supermolecular structure. The VirB4 and VirB11 ATPases and the VirD4 coupling protein are located predominantly at the cytoplasmic face of the bacterial inner membrane, whereas VirB6, a highly hydrophobic protein, most likely spans the inner membrane several times. VirD4 recognizes and recruits the export substrates by their positively charged export signal and directs them to the export apparatus. The VirB8 and VirB10 proteins have an N-terminal cytoplasmic domain, a transmembrane domain and a C-terminal periplasmic domain, suggesting that they span the inner membrane and partially localize to the bacterial cytoplasm and periplasm. VirB2, VirB3 and VirB5 are present at the periplasm, and the small lipoproteins VirB7 and VirB9 produce a disulfide-linked complex, which is localized to the bacterial outer membrane. At least three VirB proteins are required for assembly of the Agrobacterium T-pilus, with VirB2 being the major, and VirB5 and VirB7 the minor, components (reviewed in. The T-pilus is most likely required for the initial contact with the host cell but its function in the actual substrate transfer still needs to be determined. This figure is reproduced, with permission, from Ref. http://pmb.berkeley.edu/
Brief summary is given of what is known of the proteins that forms the type IV secretion apparatus of A. tumefaciens.
Virulence in A. tumefaciens is caused by the transfer of the genetic material contained in a region of the Ti plasmid called the T-DNA into plant cell. The Ti plasmid contains at least one region, virB, which encodes virulence proteins is the structural subunit of the pilus. VirB7 and VirB9 interact through a disulfide bridge and formation of a VirB7-VirB9 complex appears to be important for stabilization of many VirB proteins, notably VirB10, within the machinery; Spudich et al., 1996). VirB10 appears to self-associate within the membrane to form larger complexes. VirB4 and VirB11 have adenosine triphosphatase (ATPase) activities. VirB4 is tightly associated with the inner membrane, while VirB11 is mostly cytoplasmic and associates weakly with the inner membrane which forms the so-called type IV secretion system. This apparatus actively transfers the T-DNA of the Ti plasmid into plant cells through a pilus appendage. VirB2 is the structural subunit of the pilus. VirB7 and VirB9 interact through a disulfide bridge and formation of a VirB7-VirB9 complex appears to be important for stabilization of many VirB proteins, notably VirB10, within the machinery VirB10 appears to self-associate within the membrane to form larger complexes. VirB4 and VirB11 have adenosine triphosphatase (ATPase) activities. VirB4 is tightly associated with the inner membrane, while VirB11 is mostly cytoplasmic and associates weakly with the inner membrane.
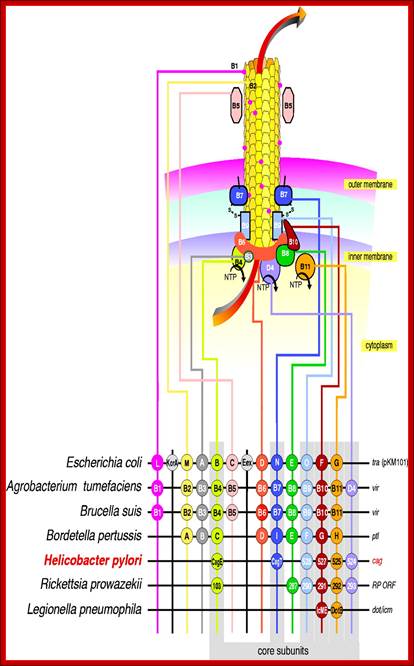
http://igem-victoria.pbworks.com/
T4SS system (repeat):
Many components of the type IV secretion system of Agrobacterium tumefaciens are homologous to proteins implicated in conjugative transfer of plasmid or phage DNAs, or in transport of toxic or transforming virulence proteins produced by bacterial pathogens targeting humans. For example, conjugative transfer of the E.coli plasmid pKM101 is carried out by a secretion apparatus, the components of which are encoded by the tra gene cluster. As shown in the figure above, many of the tra genes are homologous to the A. tumefaciens virB genes. Another example is the transport of the transforming protein CagA from Helicobacter pylori into the gastric epithelium by a secretion apparatus encoded by the cag gene cluster (see figure above). Here also, a number of the cag genes products share homology with some of the VirB proteins of Agrobacterium type IV secretion system. These observations suggest that the transport apparatus for conjugative transfer of DNA or for protein transport may consist of an Agrobacterium-like type IV secretion system.
In an attempt to crystallize all components of the type IV secretion system, we have initiated a targeted structural genomics program which aims at cloning and expressing genes encoding protein homologues of the type IV secretion system across species. In the past, crystallographers have "jumped" species very often in order to find a homologue of the protein under investigation that would yield usable crystals.
Hence, when the protein under investigation does not yield crystals or yields crystals of poor quality, it has been a common practice to renew similar attempts using a homologue of that protein using another species as a source. We applied this strategy on a small scale for a component of the type IV secretion system, the VirB11 homologue. The VirB11 homologue of H. pylori is called HP0525, while the VirB11 homologue in the plasmid RP4 conjugation system is called TrbB. While A. tumefaciens VirB11 is insoluble, HP0525 and TrbB yielded soluble and monodispersed protein preparations. However, only HP0525 crystallized. We will expand on this strategy by cloning all VirB gene homologues across 8 systems: A. tumefaciens, E. coli plasmid RP4 and pKM101 conjugal systems, H. pylori (associated with gastric and duodenal ulcers and gastric cancer), Bordetella pertussis (responsible for causing the whopping cough in infants), Rickettsia prowazeki (the causative agent for epidemic typhus), Legionella pneumophila (responsible for Legionnaires' pneumonia), and Brucella suis (the causative agent of Brucellosis in mammals), (Christie et al., 1989; Krause et al., 2000a; Krause et al., 2000b; Rashkova et al., 1997), (Zupan and Zambryski, 1995), (Eisenbrandt et al., 1999), (Anderson et al., 1996; Das et al., 1997; Fernandez et al., 1996, (Berger and Christie, 1993; Okamoto et al., 1991; Stephens et al., 1995), (Beaupre et al., 1997), (Anderson et al., 1996; Das et al., 1997; Fernandez et al., 1996; Spudich et al., 1996), (Eisenbrandt et al., 1999), (Beaupre et al., 1997),